Tianyi Zhang, William G. Whitehurst, Matthew V. Pecoraro, Junho Kim, Stefan G. Koenig and Paul J. Chirik*,
{"title":"氧化还原中性、铁介导的定向 C-H 活化:一般原理和机理启示","authors":"Tianyi Zhang, William G. Whitehurst, Matthew V. Pecoraro, Junho Kim, Stefan G. Koenig and Paul J. Chirik*, ","doi":"10.1021/jacs.4c1232910.1021/jacs.4c12329","DOIUrl":null,"url":null,"abstract":"<p >Experimental and computational studies have been conducted and established the general principles for enabling redox-neutral C–H activation by iron(II) complexes. The idealized octahedral iron(II) dimethyl complex, (depe)<sub>2</sub>Fe(CH<sub>3</sub>)<sub>2</sub> (depe = 1,2-bis(diethylphosphino)ethane) promoted the directed, regioselective <i>ortho</i> C(sp<sup>2</sup>)–H methylation of pivalophenone. The rate of the iron(II)-mediated C(sp<sup>2</sup>)–H functionalization depended on the lability of L-type phosphine ligands, the spin state of the iron center, and the size of the X-type ligands (halide, hydrocarbyl) in P<sub>4</sub>Fe<sup>II</sup>X<sub>2</sub> complexes. The C(sp<sup>2</sup>)–H alkylation reaction proved general among multiple substrates with directing groups including carbonyl, imines and pyridines. Among these, ketones and aldehydes were identified as optimal and were compatible with various steric environments and presence of acidic α-hydrogens. With stronger nitrogen donors, higher barriers for product-forming reductive elimination were observed. The effect of orbital hybridization on the chemoselectivity of C–H activation through a σ-CAM pathway by <i>d</i><sup><i>n</i>>0</sup> transition metals was also established by studying the stoichiometric reactivity of the differentially substituted (depe)<sub>2</sub>Fe(Me)R complexes (R = alkyl, aryl), where the Fe–R bond with greater <i>s</i>-character preferentially promoted selective C–H activation. Deuterium labeling and kinetic studies, coupled with computational analysis, supported a pathway involving phosphine dissociation and rate-determining C–H bond activation, leading to the observed products.</p>","PeriodicalId":49,"journal":{"name":"Journal of the American Chemical Society","volume":"146 44","pages":"30637–30652 30637–30652"},"PeriodicalIF":14.4000,"publicationDate":"2024-10-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Redox-Neutral, Iron-Mediated Directed C–H Activation: General Principles and Mechanistic Insights\",\"authors\":\"Tianyi Zhang, William G. Whitehurst, Matthew V. Pecoraro, Junho Kim, Stefan G. Koenig and Paul J. Chirik*, \",\"doi\":\"10.1021/jacs.4c1232910.1021/jacs.4c12329\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Experimental and computational studies have been conducted and established the general principles for enabling redox-neutral C–H activation by iron(II) complexes. The idealized octahedral iron(II) dimethyl complex, (depe)<sub>2</sub>Fe(CH<sub>3</sub>)<sub>2</sub> (depe = 1,2-bis(diethylphosphino)ethane) promoted the directed, regioselective <i>ortho</i> C(sp<sup>2</sup>)–H methylation of pivalophenone. The rate of the iron(II)-mediated C(sp<sup>2</sup>)–H functionalization depended on the lability of L-type phosphine ligands, the spin state of the iron center, and the size of the X-type ligands (halide, hydrocarbyl) in P<sub>4</sub>Fe<sup>II</sup>X<sub>2</sub> complexes. The C(sp<sup>2</sup>)–H alkylation reaction proved general among multiple substrates with directing groups including carbonyl, imines and pyridines. Among these, ketones and aldehydes were identified as optimal and were compatible with various steric environments and presence of acidic α-hydrogens. With stronger nitrogen donors, higher barriers for product-forming reductive elimination were observed. The effect of orbital hybridization on the chemoselectivity of C–H activation through a σ-CAM pathway by <i>d</i><sup><i>n</i>>0</sup> transition metals was also established by studying the stoichiometric reactivity of the differentially substituted (depe)<sub>2</sub>Fe(Me)R complexes (R = alkyl, aryl), where the Fe–R bond with greater <i>s</i>-character preferentially promoted selective C–H activation. Deuterium labeling and kinetic studies, coupled with computational analysis, supported a pathway involving phosphine dissociation and rate-determining C–H bond activation, leading to the observed products.</p>\",\"PeriodicalId\":49,\"journal\":{\"name\":\"Journal of the American Chemical Society\",\"volume\":\"146 44\",\"pages\":\"30637–30652 30637–30652\"},\"PeriodicalIF\":14.4000,\"publicationDate\":\"2024-10-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of the American Chemical Society\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/jacs.4c12329\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the American Chemical Society","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/jacs.4c12329","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Redox-Neutral, Iron-Mediated Directed C–H Activation: General Principles and Mechanistic Insights
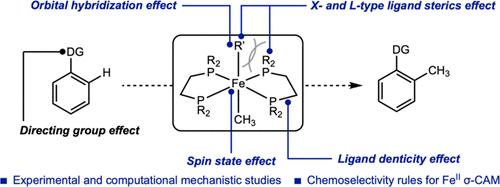
Experimental and computational studies have been conducted and established the general principles for enabling redox-neutral C–H activation by iron(II) complexes. The idealized octahedral iron(II) dimethyl complex, (depe)2Fe(CH3)2 (depe = 1,2-bis(diethylphosphino)ethane) promoted the directed, regioselective ortho C(sp2)–H methylation of pivalophenone. The rate of the iron(II)-mediated C(sp2)–H functionalization depended on the lability of L-type phosphine ligands, the spin state of the iron center, and the size of the X-type ligands (halide, hydrocarbyl) in P4FeIIX2 complexes. The C(sp2)–H alkylation reaction proved general among multiple substrates with directing groups including carbonyl, imines and pyridines. Among these, ketones and aldehydes were identified as optimal and were compatible with various steric environments and presence of acidic α-hydrogens. With stronger nitrogen donors, higher barriers for product-forming reductive elimination were observed. The effect of orbital hybridization on the chemoselectivity of C–H activation through a σ-CAM pathway by dn>0 transition metals was also established by studying the stoichiometric reactivity of the differentially substituted (depe)2Fe(Me)R complexes (R = alkyl, aryl), where the Fe–R bond with greater s-character preferentially promoted selective C–H activation. Deuterium labeling and kinetic studies, coupled with computational analysis, supported a pathway involving phosphine dissociation and rate-determining C–H bond activation, leading to the observed products.
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
