Vitória Pinheiro Balestrini , Otávio Henrique Bezerra Pinto , Blake A. Simmons , John M. Gladden , Ricardo Henrique Krüger , Betania Ferraz Quirino
{"title":"木质素降解微生物联合体的新型细菌元基因组组装分析","authors":"Vitória Pinheiro Balestrini , Otávio Henrique Bezerra Pinto , Blake A. Simmons , John M. Gladden , Ricardo Henrique Krüger , Betania Ferraz Quirino","doi":"10.1016/j.crmicr.2024.100302","DOIUrl":null,"url":null,"abstract":"<div><div>Despite recent progress, bacterial degradation of lignin is not completely understood. To address the mechanisms that bacteria from unknown taxonomic groups use to perform lignin-monomer degradation, functional analysis of bacterial metagenome-assembled genomes from soil-derived consortia enriched for microorganisms capable of degrading lignin was performed. A total of 232 metagenome-assembled genomes were recovered. After applying quality criteria of at least 70 % genome completeness and contamination less than or equal to 10 %, 39 genomes were obtained. From these, a total of 14 genomes from bacteria of unknown classification at lower taxonomic levels (i.e., only classified to the order level or higher) were chosen for further functional analysis. A global analysis of the potential ecological functions of these bacteria was performed, followed by a detailed analysis of monolignol degradation pathways. The phylum with the highest number of genomes was Proteobacteria. The genomes presented functions consistent with soil-derived bacteria, like denitrification, with different metabolic capacities related to the sulfur, chlorine, arsenic and carbon cycles, in addition to the degradation of plant cell wall components like cellulose, hemicellulose, and lignin. The Sphingomonadales_OP 08 genome showed the greatest potential to degrade cellulose and hemicellulose, although it does not appear to be able to degrade lignin. The Actinobacteria_BY 70 genome presented the highest number of enzymes and pathways related to the degradation of monolignols; furthermore, it showed the greatest potential for aromatic ring breakage by different fission pathways. The genomes of the two Actinobacteria showed the caffeic acid pathway, an important phenolic compound presenting several biological properties, such as antimicrobial and antioxidant. To our knowledge, this is the first time this pathway has been reported in this class of bacteria.</div></div>","PeriodicalId":34305,"journal":{"name":"Current Research in Microbial Sciences","volume":"7 ","pages":"Article 100302"},"PeriodicalIF":5.8000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Analysis of novel bacterial metagenome-assembled genomes from lignin-degrading microbial consortia\",\"authors\":\"Vitória Pinheiro Balestrini , Otávio Henrique Bezerra Pinto , Blake A. Simmons , John M. Gladden , Ricardo Henrique Krüger , Betania Ferraz Quirino\",\"doi\":\"10.1016/j.crmicr.2024.100302\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Despite recent progress, bacterial degradation of lignin is not completely understood. To address the mechanisms that bacteria from unknown taxonomic groups use to perform lignin-monomer degradation, functional analysis of bacterial metagenome-assembled genomes from soil-derived consortia enriched for microorganisms capable of degrading lignin was performed. A total of 232 metagenome-assembled genomes were recovered. After applying quality criteria of at least 70 % genome completeness and contamination less than or equal to 10 %, 39 genomes were obtained. From these, a total of 14 genomes from bacteria of unknown classification at lower taxonomic levels (i.e., only classified to the order level or higher) were chosen for further functional analysis. A global analysis of the potential ecological functions of these bacteria was performed, followed by a detailed analysis of monolignol degradation pathways. The phylum with the highest number of genomes was Proteobacteria. The genomes presented functions consistent with soil-derived bacteria, like denitrification, with different metabolic capacities related to the sulfur, chlorine, arsenic and carbon cycles, in addition to the degradation of plant cell wall components like cellulose, hemicellulose, and lignin. The Sphingomonadales_OP 08 genome showed the greatest potential to degrade cellulose and hemicellulose, although it does not appear to be able to degrade lignin. The Actinobacteria_BY 70 genome presented the highest number of enzymes and pathways related to the degradation of monolignols; furthermore, it showed the greatest potential for aromatic ring breakage by different fission pathways. The genomes of the two Actinobacteria showed the caffeic acid pathway, an important phenolic compound presenting several biological properties, such as antimicrobial and antioxidant. To our knowledge, this is the first time this pathway has been reported in this class of bacteria.</div></div>\",\"PeriodicalId\":34305,\"journal\":{\"name\":\"Current Research in Microbial Sciences\",\"volume\":\"7 \",\"pages\":\"Article 100302\"},\"PeriodicalIF\":5.8000,\"publicationDate\":\"2024-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Current Research in Microbial Sciences\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2666517424000853\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current Research in Microbial Sciences","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2666517424000853","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
Analysis of novel bacterial metagenome-assembled genomes from lignin-degrading microbial consortia
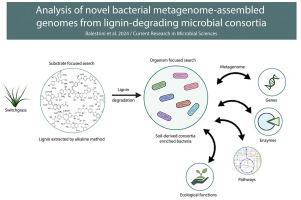
Despite recent progress, bacterial degradation of lignin is not completely understood. To address the mechanisms that bacteria from unknown taxonomic groups use to perform lignin-monomer degradation, functional analysis of bacterial metagenome-assembled genomes from soil-derived consortia enriched for microorganisms capable of degrading lignin was performed. A total of 232 metagenome-assembled genomes were recovered. After applying quality criteria of at least 70 % genome completeness and contamination less than or equal to 10 %, 39 genomes were obtained. From these, a total of 14 genomes from bacteria of unknown classification at lower taxonomic levels (i.e., only classified to the order level or higher) were chosen for further functional analysis. A global analysis of the potential ecological functions of these bacteria was performed, followed by a detailed analysis of monolignol degradation pathways. The phylum with the highest number of genomes was Proteobacteria. The genomes presented functions consistent with soil-derived bacteria, like denitrification, with different metabolic capacities related to the sulfur, chlorine, arsenic and carbon cycles, in addition to the degradation of plant cell wall components like cellulose, hemicellulose, and lignin. The Sphingomonadales_OP 08 genome showed the greatest potential to degrade cellulose and hemicellulose, although it does not appear to be able to degrade lignin. The Actinobacteria_BY 70 genome presented the highest number of enzymes and pathways related to the degradation of monolignols; furthermore, it showed the greatest potential for aromatic ring breakage by different fission pathways. The genomes of the two Actinobacteria showed the caffeic acid pathway, an important phenolic compound presenting several biological properties, such as antimicrobial and antioxidant. To our knowledge, this is the first time this pathway has been reported in this class of bacteria.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
