{"title":"1:1 卤代烃复合物中红移和蓝移氢键的比较分析","authors":"Giridhar Baburao, Aishwaryavela Esakkimuthu, Gopi Ragupathy","doi":"10.1016/j.comptc.2024.114935","DOIUrl":null,"url":null,"abstract":"<div><div>The aim of our study is to provide a possible insight into blue-shifting and red-shifting hydrogen bonding complexes. For that, we have taken CHX<sub>3</sub> (X <span><math><mo>=</mo></math></span> F, Cl, Br) with hydrogen bond acceptors such as H<sub>2</sub>O, HCl, HCN, C<sub>2</sub>H<sub>2</sub>, NH<sub>3</sub>, H<sub>2</sub>S, PH<sub>3</sub>, CH<sub>3</sub>OH, C<sub>6</sub>H<sub>6</sub> (Y <span><math><mo>=</mo></math></span> O, N, Cl, P, S, <span><math><mi>π</mi></math></span>). In this work, we have optimized and computed the vibrational frequency by performing quantum chemical calculations on the systems utilizing B3LYP and MP2 levels of theory with 6-311++G(d, p) and aug-cc-pVDZ basis sets. Further, we compared the various calculated findings, such as geometrical parameters, interaction energies (<span><math><mi>Δ</mi></math></span>E), hyper-conjugative interactions, second-order perturbation energies (E<sub>2</sub>), Laplacian electron densities (<span><math><mrow><msup><mrow><mo>∇</mo></mrow><mrow><mn>2</mn></mrow></msup><mi>ρ</mi></mrow></math></span>) at the intermolecular bond critical point (BCP), vibrational shift (<span><math><mrow><mi>Δ</mi><mi>ν</mi></mrow></math></span>). The dependence of vibrational frequency on bond length and Mulliken charge was studied. Since NH<sub>3</sub> has a highly negative Mulliken charge, it attracts the hydrogen (H) of C<img>H having a positive Mulliken charge. This effect weakens the C<img>H bond, leading to a red shift in the stretching frequency. In these complexes, NBO analysis revealed a higher value of second-order perturbation as a result of hyper-conjugative interaction from Lewis base to <span><math><msup><mrow><mi>σ</mi></mrow><mrow><mo>∗</mo></mrow></msup></math></span>(C<img>H), thus a higher redshift is observed.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1242 ","pages":"Article 114935"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Comparative analysis of red and blue-shifting hydrogen bonds in 1:1 haloform complexes\",\"authors\":\"Giridhar Baburao, Aishwaryavela Esakkimuthu, Gopi Ragupathy\",\"doi\":\"10.1016/j.comptc.2024.114935\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>The aim of our study is to provide a possible insight into blue-shifting and red-shifting hydrogen bonding complexes. For that, we have taken CHX<sub>3</sub> (X <span><math><mo>=</mo></math></span> F, Cl, Br) with hydrogen bond acceptors such as H<sub>2</sub>O, HCl, HCN, C<sub>2</sub>H<sub>2</sub>, NH<sub>3</sub>, H<sub>2</sub>S, PH<sub>3</sub>, CH<sub>3</sub>OH, C<sub>6</sub>H<sub>6</sub> (Y <span><math><mo>=</mo></math></span> O, N, Cl, P, S, <span><math><mi>π</mi></math></span>). In this work, we have optimized and computed the vibrational frequency by performing quantum chemical calculations on the systems utilizing B3LYP and MP2 levels of theory with 6-311++G(d, p) and aug-cc-pVDZ basis sets. Further, we compared the various calculated findings, such as geometrical parameters, interaction energies (<span><math><mi>Δ</mi></math></span>E), hyper-conjugative interactions, second-order perturbation energies (E<sub>2</sub>), Laplacian electron densities (<span><math><mrow><msup><mrow><mo>∇</mo></mrow><mrow><mn>2</mn></mrow></msup><mi>ρ</mi></mrow></math></span>) at the intermolecular bond critical point (BCP), vibrational shift (<span><math><mrow><mi>Δ</mi><mi>ν</mi></mrow></math></span>). The dependence of vibrational frequency on bond length and Mulliken charge was studied. Since NH<sub>3</sub> has a highly negative Mulliken charge, it attracts the hydrogen (H) of C<img>H having a positive Mulliken charge. This effect weakens the C<img>H bond, leading to a red shift in the stretching frequency. In these complexes, NBO analysis revealed a higher value of second-order perturbation as a result of hyper-conjugative interaction from Lewis base to <span><math><msup><mrow><mi>σ</mi></mrow><mrow><mo>∗</mo></mrow></msup></math></span>(C<img>H), thus a higher redshift is observed.</div></div>\",\"PeriodicalId\":284,\"journal\":{\"name\":\"Computational and Theoretical Chemistry\",\"volume\":\"1242 \",\"pages\":\"Article 114935\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computational and Theoretical Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2210271X24004742\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/29 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24004742","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/29 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Comparative analysis of red and blue-shifting hydrogen bonds in 1:1 haloform complexes
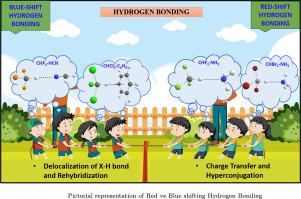
The aim of our study is to provide a possible insight into blue-shifting and red-shifting hydrogen bonding complexes. For that, we have taken CHX3 (X F, Cl, Br) with hydrogen bond acceptors such as H2O, HCl, HCN, C2H2, NH3, H2S, PH3, CH3OH, C6H6 (Y O, N, Cl, P, S, ). In this work, we have optimized and computed the vibrational frequency by performing quantum chemical calculations on the systems utilizing B3LYP and MP2 levels of theory with 6-311++G(d, p) and aug-cc-pVDZ basis sets. Further, we compared the various calculated findings, such as geometrical parameters, interaction energies (E), hyper-conjugative interactions, second-order perturbation energies (E2), Laplacian electron densities () at the intermolecular bond critical point (BCP), vibrational shift (). The dependence of vibrational frequency on bond length and Mulliken charge was studied. Since NH3 has a highly negative Mulliken charge, it attracts the hydrogen (H) of CH having a positive Mulliken charge. This effect weakens the CH bond, leading to a red shift in the stretching frequency. In these complexes, NBO analysis revealed a higher value of second-order perturbation as a result of hyper-conjugative interaction from Lewis base to (CH), thus a higher redshift is observed.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
