{"title":"DC24: 用于多配置密度相干函数理论的新密度相干函数","authors":"Dayou Zhang, Yinan Shu, Donald G. Truhlar","doi":"10.1002/jcc.27522","DOIUrl":null,"url":null,"abstract":"In this study, we explored several alternative functional forms to construct more accurate and more physical density coherence (DC) functionals for multiconfiguration density‐coherence functional theory. Each functional is parameterized against the same database as used in our previous work. The best DC functional, which is called DC24, has a more physical interpretation, and—as a side benefit—it also has a mean unsigned error of 1.73 kcal/mol, which is a 9% improvement as compared to the previous functional. The article also contains a new definition of the unpaired electron density, which may be useful in other contexts as well.","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"18 1","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-11-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"DC24: A new density coherence functional for multiconfiguration density‐coherence functional theory\",\"authors\":\"Dayou Zhang, Yinan Shu, Donald G. Truhlar\",\"doi\":\"10.1002/jcc.27522\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"In this study, we explored several alternative functional forms to construct more accurate and more physical density coherence (DC) functionals for multiconfiguration density‐coherence functional theory. Each functional is parameterized against the same database as used in our previous work. The best DC functional, which is called DC24, has a more physical interpretation, and—as a side benefit—it also has a mean unsigned error of 1.73 kcal/mol, which is a 9% improvement as compared to the previous functional. The article also contains a new definition of the unpaired electron density, which may be useful in other contexts as well.\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"18 1\",\"pages\":\"\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-11-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1002/jcc.27522\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1002/jcc.27522","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
摘要
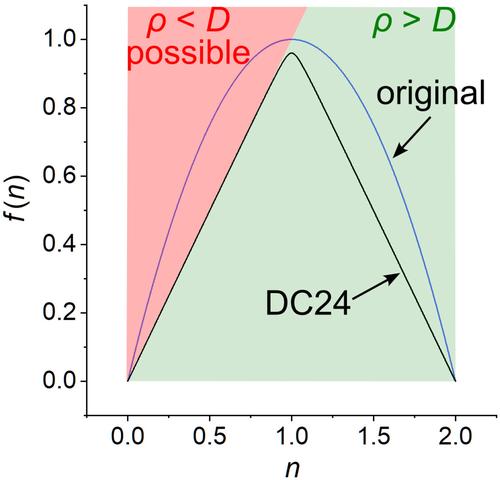
在这项研究中,我们探索了几种可供选择的函数形式,以便为多配置密度相干函数理论构建更精确、更物理的密度相干(DC)函数。每个函数的参数都与我们之前工作中使用的数据库相同。最好的 DC 函数被称为 DC24,它有更多的物理解释,同时它的平均无符号误差为 1.73 kcal/mol,比之前的函数提高了 9%。文章还包含了未配对电子密度的新定义,这在其他情况下也可能有用。
DC24: A new density coherence functional for multiconfiguration density‐coherence functional theory
In this study, we explored several alternative functional forms to construct more accurate and more physical density coherence (DC) functionals for multiconfiguration density‐coherence functional theory. Each functional is parameterized against the same database as used in our previous work. The best DC functional, which is called DC24, has a more physical interpretation, and—as a side benefit—it also has a mean unsigned error of 1.73 kcal/mol, which is a 9% improvement as compared to the previous functional. The article also contains a new definition of the unpaired electron density, which may be useful in other contexts as well.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
