Youngbin Moon, Christina J Herrmann, Aleksei Mironov, Mihaela Zavolan
{"title":"PolyASite v3.0:从单细胞 RNA 序列数据推断出的多腺苷酸化位点多物种图谱。","authors":"Youngbin Moon, Christina J Herrmann, Aleksei Mironov, Mihaela Zavolan","doi":"10.1093/nar/gkae1043","DOIUrl":null,"url":null,"abstract":"<p><p>The broadly used 10X Genomics technology for single-cell RNA sequencing (scRNA-seq) captures RNA 3' ends. Thus, some reads contain part of the non-templated polyadenosine tails, providing direct evidence for the sites of 3' end cleavage and polyadenylation on the respective RNAs. Taking advantage of this property, we recently developed the SCINPAS workflow to infer polyadenylation sites (PASs) from scRNA-seq data. Here, we used this workflow to construct version 3.0 (v3.0, https://polyasite.unibas.ch/) of the PolyASite Atlas from a big compendium of publicly available human, mouse and worm scRNA-seq datasets obtained from healthy tissues. As the resolution of scRNA-seq was too low for robust detection of cell-level differences in PAS usage, we aggregated samples based on their tissue-of-origin to construct tissue-level catalogs of PASs. These provide qualitatively new information about PAS usage, in comparison to the previous PAS catalogs that were based on bulk 3' end sequencing experiments primarily in cell lines. In the new version, we document stringency levels associated with each PAS so that users can balance sensitivity and specificity in their analysis. We also upgraded the integration with the UCSC Genome Browser and developed track hubs conveniently displaying pooled and tissue-specific expression of PASs.</p>","PeriodicalId":19471,"journal":{"name":"Nucleic Acids Research","volume":" ","pages":"D197-D204"},"PeriodicalIF":15.0000,"publicationDate":"2025-01-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11701536/pdf/","citationCount":"0","resultStr":"{\"title\":\"PolyASite v3.0: a multi-species atlas of polyadenylation sites inferred from single-cell RNA-sequencing data.\",\"authors\":\"Youngbin Moon, Christina J Herrmann, Aleksei Mironov, Mihaela Zavolan\",\"doi\":\"10.1093/nar/gkae1043\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The broadly used 10X Genomics technology for single-cell RNA sequencing (scRNA-seq) captures RNA 3' ends. Thus, some reads contain part of the non-templated polyadenosine tails, providing direct evidence for the sites of 3' end cleavage and polyadenylation on the respective RNAs. Taking advantage of this property, we recently developed the SCINPAS workflow to infer polyadenylation sites (PASs) from scRNA-seq data. Here, we used this workflow to construct version 3.0 (v3.0, https://polyasite.unibas.ch/) of the PolyASite Atlas from a big compendium of publicly available human, mouse and worm scRNA-seq datasets obtained from healthy tissues. As the resolution of scRNA-seq was too low for robust detection of cell-level differences in PAS usage, we aggregated samples based on their tissue-of-origin to construct tissue-level catalogs of PASs. These provide qualitatively new information about PAS usage, in comparison to the previous PAS catalogs that were based on bulk 3' end sequencing experiments primarily in cell lines. In the new version, we document stringency levels associated with each PAS so that users can balance sensitivity and specificity in their analysis. We also upgraded the integration with the UCSC Genome Browser and developed track hubs conveniently displaying pooled and tissue-specific expression of PASs.</p>\",\"PeriodicalId\":19471,\"journal\":{\"name\":\"Nucleic Acids Research\",\"volume\":\" \",\"pages\":\"D197-D204\"},\"PeriodicalIF\":15.0000,\"publicationDate\":\"2025-01-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11701536/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nucleic Acids Research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/nar/gkae1043\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nucleic Acids Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/nar/gkae1043","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
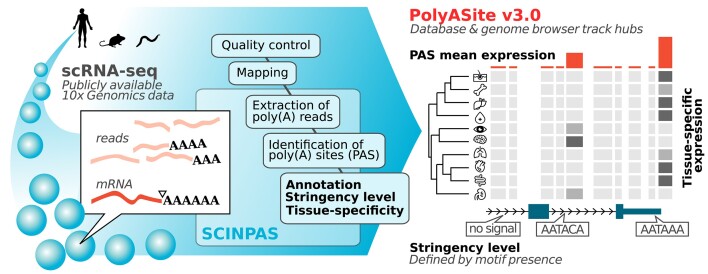
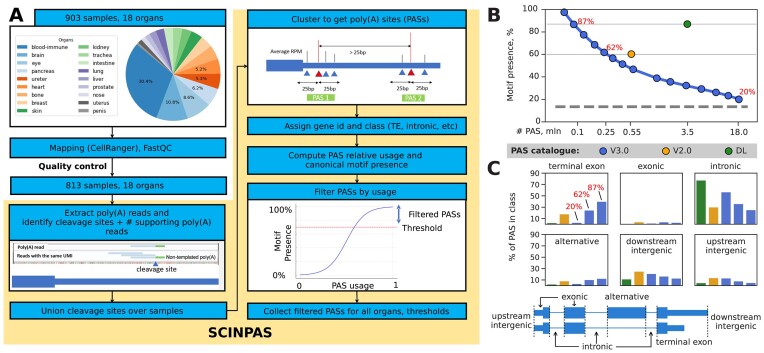
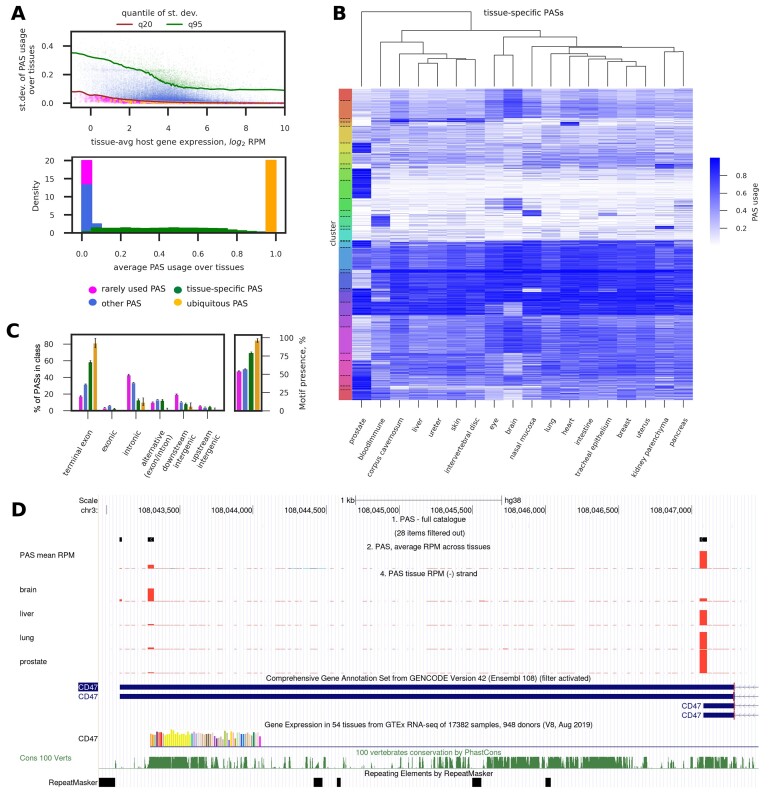
广泛使用的 10X Genomics 单细胞 RNA 测序(scRNA-seq)技术可捕获 RNA 3' 端。因此,一些读数包含了部分非模板多聚腺苷尾,为相应 RNA 的 3' 端裂解和多聚腺苷化位点提供了直接证据。利用这一特性,我们最近开发了 SCINPAS 工作流程,从 scRNA-seq 数据中推断多腺苷酸化位点(PAS)。在这里,我们利用这一工作流程,从从健康组织中获取的大量公开可用的人类、小鼠和蠕虫scRNA-seq数据集中构建了PolyASite Atlas的3.0版本(v3.0,https://polyasite.unibas.ch/)。由于 scRNA-seq 的分辨率太低,无法稳健地检测 PAS 使用的细胞级差异,因此我们根据样本的原生组织对样本进行了汇总,以构建组织级的 PAS 目录。与之前基于主要在细胞系中进行的大量 3' 端测序实验的 PAS 目录相比,这些目录提供了有关 PAS 使用情况的新的定性信息。在新版本中,我们记录了与每个 PAS 相关的严格程度,以便用户在分析中平衡灵敏度和特异性。我们还升级了与 UCSC 基因组浏览器的整合,并开发了跟踪中心,方便显示 PAS 的集合表达和组织特异性表达。
PolyASite v3.0: a multi-species atlas of polyadenylation sites inferred from single-cell RNA-sequencing data.
The broadly used 10X Genomics technology for single-cell RNA sequencing (scRNA-seq) captures RNA 3' ends. Thus, some reads contain part of the non-templated polyadenosine tails, providing direct evidence for the sites of 3' end cleavage and polyadenylation on the respective RNAs. Taking advantage of this property, we recently developed the SCINPAS workflow to infer polyadenylation sites (PASs) from scRNA-seq data. Here, we used this workflow to construct version 3.0 (v3.0, https://polyasite.unibas.ch/) of the PolyASite Atlas from a big compendium of publicly available human, mouse and worm scRNA-seq datasets obtained from healthy tissues. As the resolution of scRNA-seq was too low for robust detection of cell-level differences in PAS usage, we aggregated samples based on their tissue-of-origin to construct tissue-level catalogs of PASs. These provide qualitatively new information about PAS usage, in comparison to the previous PAS catalogs that were based on bulk 3' end sequencing experiments primarily in cell lines. In the new version, we document stringency levels associated with each PAS so that users can balance sensitivity and specificity in their analysis. We also upgraded the integration with the UCSC Genome Browser and developed track hubs conveniently displaying pooled and tissue-specific expression of PASs.
期刊介绍:
Nucleic Acids Research (NAR) is a scientific journal that publishes research on various aspects of nucleic acids and proteins involved in nucleic acid metabolism and interactions. It covers areas such as chemistry and synthetic biology, computational biology, gene regulation, chromatin and epigenetics, genome integrity, repair and replication, genomics, molecular biology, nucleic acid enzymes, RNA, and structural biology. The journal also includes a Survey and Summary section for brief reviews. Additionally, each year, the first issue is dedicated to biological databases, and an issue in July focuses on web-based software resources for the biological community. Nucleic Acids Research is indexed by several services including Abstracts on Hygiene and Communicable Diseases, Animal Breeding Abstracts, Agricultural Engineering Abstracts, Agbiotech News and Information, BIOSIS Previews, CAB Abstracts, and EMBASE.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
