Hossein Shirani*, and , Seyed Majid Hashemianzadeh*,
{"title":"机器学习预测白藜芦醇药物的势能面:量子级计算","authors":"Hossein Shirani*, and , Seyed Majid Hashemianzadeh*, ","doi":"10.1021/acsmedchemlett.4c0038210.1021/acsmedchemlett.4c00382","DOIUrl":null,"url":null,"abstract":"<p >The ANI-1x neural network potential, trained on the density functional theory data set, as a quantum-level machine learning calculation has been investigated to forecast the potential energy surfaces of the Resveratrol (3,5,4′-trihydroxy-<i>trans</i>-stilbene) antiparkinsonian drug in a very short computing time. A comprehensive validation of the ANI-1x deep learning technique was provided on the Resveratrol molecule using density functional theory at the wB97X/6-31G(d) level of theory. The results showcased in this study will offer significant insights into pharmaceutical computational research, medicinal chemistry, drug discovery and design, thereby making a valuable contribution.</p>","PeriodicalId":20,"journal":{"name":"ACS Medicinal Chemistry Letters","volume":"15 11","pages":"1979–1986 1979–1986"},"PeriodicalIF":3.5000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Machine Learning to Predict Potential Energy Surface of Resveratrol Drug: A Quantum-Level Calculation\",\"authors\":\"Hossein Shirani*, and , Seyed Majid Hashemianzadeh*, \",\"doi\":\"10.1021/acsmedchemlett.4c0038210.1021/acsmedchemlett.4c00382\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The ANI-1x neural network potential, trained on the density functional theory data set, as a quantum-level machine learning calculation has been investigated to forecast the potential energy surfaces of the Resveratrol (3,5,4′-trihydroxy-<i>trans</i>-stilbene) antiparkinsonian drug in a very short computing time. A comprehensive validation of the ANI-1x deep learning technique was provided on the Resveratrol molecule using density functional theory at the wB97X/6-31G(d) level of theory. The results showcased in this study will offer significant insights into pharmaceutical computational research, medicinal chemistry, drug discovery and design, thereby making a valuable contribution.</p>\",\"PeriodicalId\":20,\"journal\":{\"name\":\"ACS Medicinal Chemistry Letters\",\"volume\":\"15 11\",\"pages\":\"1979–1986 1979–1986\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Medicinal Chemistry Letters\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acsmedchemlett.4c00382\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Medicinal Chemistry Letters","FirstCategoryId":"3","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acsmedchemlett.4c00382","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Machine Learning to Predict Potential Energy Surface of Resveratrol Drug: A Quantum-Level Calculation
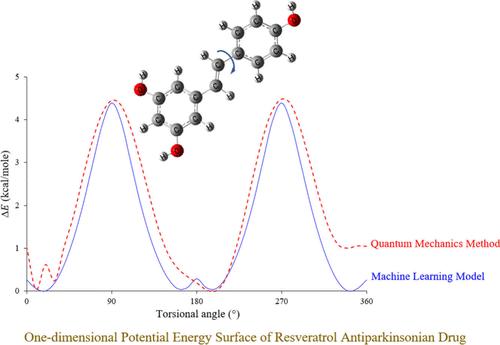
The ANI-1x neural network potential, trained on the density functional theory data set, as a quantum-level machine learning calculation has been investigated to forecast the potential energy surfaces of the Resveratrol (3,5,4′-trihydroxy-trans-stilbene) antiparkinsonian drug in a very short computing time. A comprehensive validation of the ANI-1x deep learning technique was provided on the Resveratrol molecule using density functional theory at the wB97X/6-31G(d) level of theory. The results showcased in this study will offer significant insights into pharmaceutical computational research, medicinal chemistry, drug discovery and design, thereby making a valuable contribution.
期刊介绍:
ACS Medicinal Chemistry Letters is interested in receiving manuscripts that discuss various aspects of medicinal chemistry. The journal will publish studies that pertain to a broad range of subject matter, including compound design and optimization, biological evaluation, drug delivery, imaging agents, and pharmacology of both small and large bioactive molecules. Specific areas include but are not limited to:
Identification, synthesis, and optimization of lead biologically active molecules and drugs (small molecules and biologics)
Biological characterization of new molecular entities in the context of drug discovery
Computational, cheminformatics, and structural studies for the identification or SAR analysis of bioactive molecules, ligands and their targets, etc.
Novel and improved methodologies, including radiation biochemistry, with broad application to medicinal chemistry
Discovery technologies for biologically active molecules from both synthetic and natural (plant and other) sources
Pharmacokinetic/pharmacodynamic studies that address mechanisms underlying drug disposition and response
Pharmacogenetic and pharmacogenomic studies used to enhance drug design and the translation of medicinal chemistry into the clinic
Mechanistic drug metabolism and regulation of metabolic enzyme gene expression
Chemistry patents relevant to the medicinal chemistry field.


 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
