{"title":"MPA-MutPred:准确预测膜蛋白复合物突变时结合亲和力变化的新策略。","authors":"Fathima Ridha, M Michael Gromiha","doi":"10.1093/bib/bbae598","DOIUrl":null,"url":null,"abstract":"<p><p>Mutations in the interface of membrane protein (MP) complexes are key contributors to a broad spectrum of human diseases, primarily due to changes in their binding affinities. While various methods exist for predicting the mutation-induced changes in binding affinity (ΔΔG) in protein-protein complexes, none are specific to MP complexes. This study proposes a novel strategy for ΔΔG prediction in MP complexes, which combines linear and nonlinear models, to obtain a more robust model with improved prediction accuracy. We used multiple linear regression to extract informative features that influence the binding affinity in MP complexes, which included changes in the stability of the complex, conservation score, electrostatic interaction, relatively accessible surface area, and interface contacts. Further, using gradient boosting regressor on the selected features, we developed MPA-MutPred, a novel method specific for predicting the ΔΔG of membrane protein-protein complexes, and it is freely accessible at https://web.iitm.ac.in/bioinfo2/MPA-MutPred/. Our method achieved a correlation of 0.75 and a mean absolute error (MAE) of 0.73 kcal/mol in the jack-knife test conducted on a dataset of 770 mutants. We further validated the method using a blind test set of 86 mutations, obtaining a correlation of 0.85 and an MAE of 0.77 kcal/mol. We anticipate that this method can be used for large-scale studies to understand the influence of binding affinity change on disease-causing mutations in MP complexes, thereby aiding in the understanding of disease mechanisms and the identification of potential therapeutic targets.</p>","PeriodicalId":9209,"journal":{"name":"Briefings in bioinformatics","volume":"25 6","pages":""},"PeriodicalIF":7.7000,"publicationDate":"2024-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11568875/pdf/","citationCount":"0","resultStr":"{\"title\":\"MPA-MutPred: a novel strategy for accurately predicting the binding affinity change upon mutation in membrane protein complexes.\",\"authors\":\"Fathima Ridha, M Michael Gromiha\",\"doi\":\"10.1093/bib/bbae598\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Mutations in the interface of membrane protein (MP) complexes are key contributors to a broad spectrum of human diseases, primarily due to changes in their binding affinities. While various methods exist for predicting the mutation-induced changes in binding affinity (ΔΔG) in protein-protein complexes, none are specific to MP complexes. This study proposes a novel strategy for ΔΔG prediction in MP complexes, which combines linear and nonlinear models, to obtain a more robust model with improved prediction accuracy. We used multiple linear regression to extract informative features that influence the binding affinity in MP complexes, which included changes in the stability of the complex, conservation score, electrostatic interaction, relatively accessible surface area, and interface contacts. Further, using gradient boosting regressor on the selected features, we developed MPA-MutPred, a novel method specific for predicting the ΔΔG of membrane protein-protein complexes, and it is freely accessible at https://web.iitm.ac.in/bioinfo2/MPA-MutPred/. Our method achieved a correlation of 0.75 and a mean absolute error (MAE) of 0.73 kcal/mol in the jack-knife test conducted on a dataset of 770 mutants. We further validated the method using a blind test set of 86 mutations, obtaining a correlation of 0.85 and an MAE of 0.77 kcal/mol. We anticipate that this method can be used for large-scale studies to understand the influence of binding affinity change on disease-causing mutations in MP complexes, thereby aiding in the understanding of disease mechanisms and the identification of potential therapeutic targets.</p>\",\"PeriodicalId\":9209,\"journal\":{\"name\":\"Briefings in bioinformatics\",\"volume\":\"25 6\",\"pages\":\"\"},\"PeriodicalIF\":7.7000,\"publicationDate\":\"2024-09-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11568875/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Briefings in bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bib/bbae598\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Briefings in bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bib/bbae598","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
MPA-MutPred: a novel strategy for accurately predicting the binding affinity change upon mutation in membrane protein complexes.
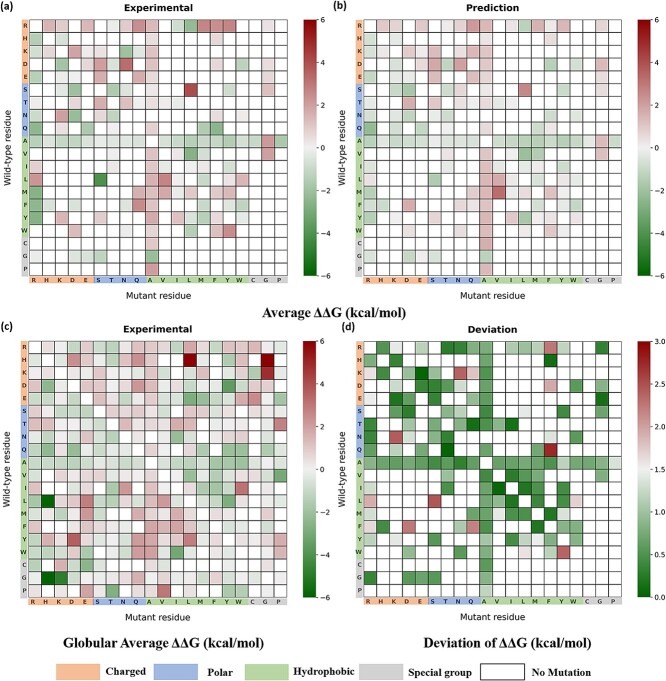
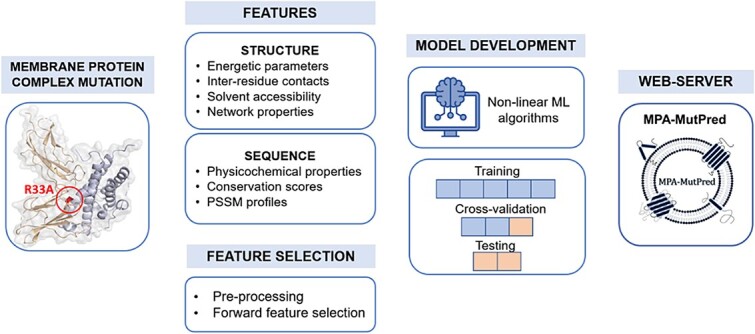
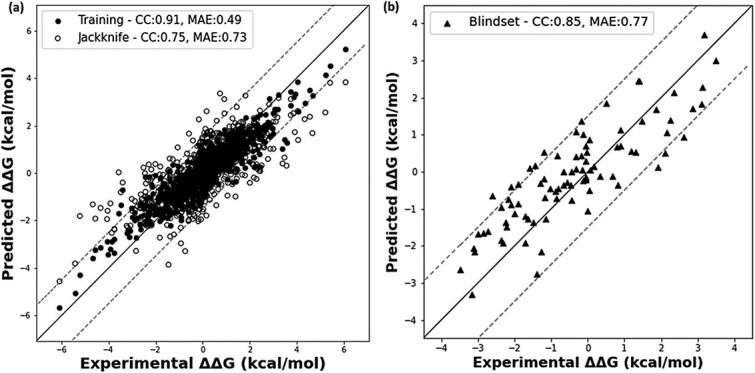
Mutations in the interface of membrane protein (MP) complexes are key contributors to a broad spectrum of human diseases, primarily due to changes in their binding affinities. While various methods exist for predicting the mutation-induced changes in binding affinity (ΔΔG) in protein-protein complexes, none are specific to MP complexes. This study proposes a novel strategy for ΔΔG prediction in MP complexes, which combines linear and nonlinear models, to obtain a more robust model with improved prediction accuracy. We used multiple linear regression to extract informative features that influence the binding affinity in MP complexes, which included changes in the stability of the complex, conservation score, electrostatic interaction, relatively accessible surface area, and interface contacts. Further, using gradient boosting regressor on the selected features, we developed MPA-MutPred, a novel method specific for predicting the ΔΔG of membrane protein-protein complexes, and it is freely accessible at https://web.iitm.ac.in/bioinfo2/MPA-MutPred/. Our method achieved a correlation of 0.75 and a mean absolute error (MAE) of 0.73 kcal/mol in the jack-knife test conducted on a dataset of 770 mutants. We further validated the method using a blind test set of 86 mutations, obtaining a correlation of 0.85 and an MAE of 0.77 kcal/mol. We anticipate that this method can be used for large-scale studies to understand the influence of binding affinity change on disease-causing mutations in MP complexes, thereby aiding in the understanding of disease mechanisms and the identification of potential therapeutic targets.
期刊介绍:
Briefings in Bioinformatics is an international journal serving as a platform for researchers and educators in the life sciences. It also appeals to mathematicians, statisticians, and computer scientists applying their expertise to biological challenges. The journal focuses on reviews tailored for users of databases and analytical tools in contemporary genetics, molecular and systems biology. It stands out by offering practical assistance and guidance to non-specialists in computerized methodologies. Covering a wide range from introductory concepts to specific protocols and analyses, the papers address bacterial, plant, fungal, animal, and human data.
The journal's detailed subject areas include genetic studies of phenotypes and genotypes, mapping, DNA sequencing, expression profiling, gene expression studies, microarrays, alignment methods, protein profiles and HMMs, lipids, metabolic and signaling pathways, structure determination and function prediction, phylogenetic studies, and education and training.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
