{"title":"利用共振增强多光子电离和慢电子速度图成像技术对 3,4-二氟苯乙炔进行振动赋值和能量分析","authors":"Zefeng Hua, Xiangxiang Wang, Jinhui Deng, Xinyan Yang, Zhongfa Sun, Xianfeng Zheng, Zhengbo Qin","doi":"10.1016/j.cplett.2024.141730","DOIUrl":null,"url":null,"abstract":"<div><div>Here, we report spectroscopic investigations of 3,4-difluorophenylacetylene applying two-color resonant two-photon ionization (2C-R2PI) and slow electron velocity-map imaging (SEVI) techniques. With the assistance of density functional theory (DFT) calculations, vibrational modes of the S<sub>0</sub> neutral ground state, S<sub>1</sub> first electronic excited state, and D<sub>0</sub> cationic ground state were assigned. The adiabatic excitation energy of the S<sub>1</sub> ← S<sub>0</sub> electronic transition was determined to be 35639 ± 5 cm<sup>−1</sup> from the resonance-enhanced multiphoton ionization (REMPI) spectroscopy. Additionally, the adiabatic ionization potential was found to be 72398 ± 5 cm<sup>−1</sup> based on the SEVI spectra.</div></div>","PeriodicalId":273,"journal":{"name":"Chemical Physics Letters","volume":"857 ","pages":"Article 141730"},"PeriodicalIF":3.1000,"publicationDate":"2024-12-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Vibrational assignments and energy analyses of 3,4-difluorophenylacetylene using resonance-enhanced multiphoton ionization and slow electron velocity-map imaging techniques\",\"authors\":\"Zefeng Hua, Xiangxiang Wang, Jinhui Deng, Xinyan Yang, Zhongfa Sun, Xianfeng Zheng, Zhengbo Qin\",\"doi\":\"10.1016/j.cplett.2024.141730\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Here, we report spectroscopic investigations of 3,4-difluorophenylacetylene applying two-color resonant two-photon ionization (2C-R2PI) and slow electron velocity-map imaging (SEVI) techniques. With the assistance of density functional theory (DFT) calculations, vibrational modes of the S<sub>0</sub> neutral ground state, S<sub>1</sub> first electronic excited state, and D<sub>0</sub> cationic ground state were assigned. The adiabatic excitation energy of the S<sub>1</sub> ← S<sub>0</sub> electronic transition was determined to be 35639 ± 5 cm<sup>−1</sup> from the resonance-enhanced multiphoton ionization (REMPI) spectroscopy. Additionally, the adiabatic ionization potential was found to be 72398 ± 5 cm<sup>−1</sup> based on the SEVI spectra.</div></div>\",\"PeriodicalId\":273,\"journal\":{\"name\":\"Chemical Physics Letters\",\"volume\":\"857 \",\"pages\":\"Article 141730\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-12-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Chemical Physics Letters\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0009261424006729\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/1 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemical Physics Letters","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0009261424006729","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/1 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Vibrational assignments and energy analyses of 3,4-difluorophenylacetylene using resonance-enhanced multiphoton ionization and slow electron velocity-map imaging techniques
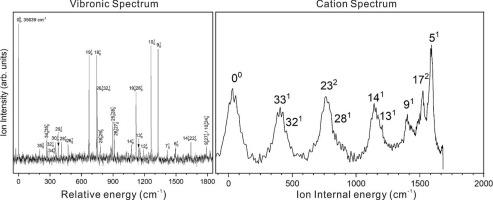
Here, we report spectroscopic investigations of 3,4-difluorophenylacetylene applying two-color resonant two-photon ionization (2C-R2PI) and slow electron velocity-map imaging (SEVI) techniques. With the assistance of density functional theory (DFT) calculations, vibrational modes of the S0 neutral ground state, S1 first electronic excited state, and D0 cationic ground state were assigned. The adiabatic excitation energy of the S1 ← S0 electronic transition was determined to be 35639 ± 5 cm−1 from the resonance-enhanced multiphoton ionization (REMPI) spectroscopy. Additionally, the adiabatic ionization potential was found to be 72398 ± 5 cm−1 based on the SEVI spectra.
期刊介绍:
Chemical Physics Letters has an open access mirror journal, Chemical Physics Letters: X, sharing the same aims and scope, editorial team, submission system and rigorous peer review.
Chemical Physics Letters publishes brief reports on molecules, interfaces, condensed phases, nanomaterials and nanostructures, polymers, biomolecular systems, and energy conversion and storage.
Criteria for publication are quality, urgency and impact. Further, experimental results reported in the journal have direct relevance for theory, and theoretical developments or non-routine computations relate directly to experiment. Manuscripts must satisfy these criteria and should not be minor extensions of previous work.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
