Neha M. Mhetre , Aniket L. Bhatambrekar , D. Priya , Venkatesan Saravanan , Muthukumaradoss Kathiravan , Krishna S. Shevate , Kalirajan Rajagopal , Kalyani D. Asgaonkar , Trupti S. Chitre
{"title":"利用QSAR、ADMET、分子对接、MM-GBSA和分子动力学模拟方法,合理设计一些1,3,4三取代吡唑-噻唑衍生物作为MtInhA抑制剂","authors":"Neha M. Mhetre , Aniket L. Bhatambrekar , D. Priya , Venkatesan Saravanan , Muthukumaradoss Kathiravan , Krishna S. Shevate , Kalirajan Rajagopal , Kalyani D. Asgaonkar , Trupti S. Chitre","doi":"10.1016/j.chphi.2024.100769","DOIUrl":null,"url":null,"abstract":"<div><div>Using computational approaches, the potential efficacy and specificity of 1,3,4 trisubstituted pyrazole derivatives as <em>Mt</em>InhA inhibitors which will aid in rational drug design for tubercular therapy were forecasted. QSARINS software was used to investigate the ability of compound to inhibit <em>Mt</em>InhA. Three noteworthy descriptors with significant correlations and impressive statistical values were identified by the produced QSAR model: Correlation of coefficient(R<sup>2</sup>)= 0.8789, Cross-validation leave one out correlation coefficient (Q<sup>2</sup>LOO)= 0.8402, Cross-validation leave-many-out correlation coefficient(Q<sup>2</sup>LMO)=0.7321, Concordance Correlation Coefficient for cross-validation(CCC<sub>tr</sub>)=0.9355, CCC<sub>ext</sub> =0.888. The descriptor generated by the QSAR model includes Centered Broto-Moreau autocorrelation weighted by Sanderson electronegativities (ATSC1e), Radial distribution functions at 15.0 and 2.0 Å inter-atomic distances weighted by relative van der Waals volumes (RDF150v), Radial distribution function – 145/weighted by relative I-state (RDF145s). Using these, three descriptor model was developed and the designed compounds were evaluated for their <em>Mt</em>InhA inhibitory activity. Further, ADMET prediction and Molecular docking studies were carried out using Schrodinger's Software. ADMET prediction were used to evaluate drug likeliness and molecular docking was used to determine the interactions of designed compounds with the target protein. After the docking studies, the compounds were subjected for MM-GBSA calculations and MD simulation. Among the designed compounds, <strong>AP2</strong> had the strongest binding affinity towards the <em>Mt</em>InhA enzyme. The result of this work helps to understand the key interactions between 1,3,4 trisubstituted pyrazole derivatives and <em>Mt</em>InhA protein that may be necessary to develop new lead compounds against tuberculosis.</div></div>","PeriodicalId":9758,"journal":{"name":"Chemical Physics Impact","volume":"9 ","pages":"Article 100769"},"PeriodicalIF":5.3000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Rational design of some 1,3,4 trisubstituted pyrazole-thiazole derivatives to serve as MtInhA inhibitors using QSAR, ADMET, molecular docking, MM-GBSA, and molecular dynamics simulations approach\",\"authors\":\"Neha M. Mhetre , Aniket L. Bhatambrekar , D. Priya , Venkatesan Saravanan , Muthukumaradoss Kathiravan , Krishna S. Shevate , Kalirajan Rajagopal , Kalyani D. Asgaonkar , Trupti S. Chitre\",\"doi\":\"10.1016/j.chphi.2024.100769\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Using computational approaches, the potential efficacy and specificity of 1,3,4 trisubstituted pyrazole derivatives as <em>Mt</em>InhA inhibitors which will aid in rational drug design for tubercular therapy were forecasted. QSARINS software was used to investigate the ability of compound to inhibit <em>Mt</em>InhA. Three noteworthy descriptors with significant correlations and impressive statistical values were identified by the produced QSAR model: Correlation of coefficient(R<sup>2</sup>)= 0.8789, Cross-validation leave one out correlation coefficient (Q<sup>2</sup>LOO)= 0.8402, Cross-validation leave-many-out correlation coefficient(Q<sup>2</sup>LMO)=0.7321, Concordance Correlation Coefficient for cross-validation(CCC<sub>tr</sub>)=0.9355, CCC<sub>ext</sub> =0.888. The descriptor generated by the QSAR model includes Centered Broto-Moreau autocorrelation weighted by Sanderson electronegativities (ATSC1e), Radial distribution functions at 15.0 and 2.0 Å inter-atomic distances weighted by relative van der Waals volumes (RDF150v), Radial distribution function – 145/weighted by relative I-state (RDF145s). Using these, three descriptor model was developed and the designed compounds were evaluated for their <em>Mt</em>InhA inhibitory activity. Further, ADMET prediction and Molecular docking studies were carried out using Schrodinger's Software. ADMET prediction were used to evaluate drug likeliness and molecular docking was used to determine the interactions of designed compounds with the target protein. After the docking studies, the compounds were subjected for MM-GBSA calculations and MD simulation. Among the designed compounds, <strong>AP2</strong> had the strongest binding affinity towards the <em>Mt</em>InhA enzyme. The result of this work helps to understand the key interactions between 1,3,4 trisubstituted pyrazole derivatives and <em>Mt</em>InhA protein that may be necessary to develop new lead compounds against tuberculosis.</div></div>\",\"PeriodicalId\":9758,\"journal\":{\"name\":\"Chemical Physics Impact\",\"volume\":\"9 \",\"pages\":\"Article 100769\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Chemical Physics Impact\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S266702242400313X\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/3 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemical Physics Impact","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S266702242400313X","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/3 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Rational design of some 1,3,4 trisubstituted pyrazole-thiazole derivatives to serve as MtInhA inhibitors using QSAR, ADMET, molecular docking, MM-GBSA, and molecular dynamics simulations approach
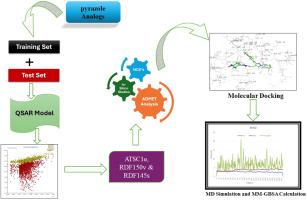
Using computational approaches, the potential efficacy and specificity of 1,3,4 trisubstituted pyrazole derivatives as MtInhA inhibitors which will aid in rational drug design for tubercular therapy were forecasted. QSARINS software was used to investigate the ability of compound to inhibit MtInhA. Three noteworthy descriptors with significant correlations and impressive statistical values were identified by the produced QSAR model: Correlation of coefficient(R2)= 0.8789, Cross-validation leave one out correlation coefficient (Q2LOO)= 0.8402, Cross-validation leave-many-out correlation coefficient(Q2LMO)=0.7321, Concordance Correlation Coefficient for cross-validation(CCCtr)=0.9355, CCCext =0.888. The descriptor generated by the QSAR model includes Centered Broto-Moreau autocorrelation weighted by Sanderson electronegativities (ATSC1e), Radial distribution functions at 15.0 and 2.0 Å inter-atomic distances weighted by relative van der Waals volumes (RDF150v), Radial distribution function – 145/weighted by relative I-state (RDF145s). Using these, three descriptor model was developed and the designed compounds were evaluated for their MtInhA inhibitory activity. Further, ADMET prediction and Molecular docking studies were carried out using Schrodinger's Software. ADMET prediction were used to evaluate drug likeliness and molecular docking was used to determine the interactions of designed compounds with the target protein. After the docking studies, the compounds were subjected for MM-GBSA calculations and MD simulation. Among the designed compounds, AP2 had the strongest binding affinity towards the MtInhA enzyme. The result of this work helps to understand the key interactions between 1,3,4 trisubstituted pyrazole derivatives and MtInhA protein that may be necessary to develop new lead compounds against tuberculosis.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
