{"title":"明尼苏达函数在振动频率上的表现","authors":"Jiaxu Wang, Cheng Zhang, Yaqi Li, Yini Zhou, Yuanyuan Shu, Songping Liang, Gaihua Zhang, Zhonghua Liu, Ying Wang","doi":"10.1002/qua.27516","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>Molecular geometry and harmonic frequency calculations are essential in thermochemical computations, with density functional theory (DFT) being widely employed for vibrational frequency predictions due to its efficiency and accuracy. In this study, we assessed the precision of 28 Minnesota based functionals with three different basis sets using the VIBFREQ1295 dataset. Scaling factors are necessary for predicting fundamental frequencies, global scaling factors were fitted by using F38/10 and VIBFREQ1295 datasets. The superior performing functionals were then fitted based on vibrational frequency ranges to obtain frequency-range-specific scaling factors. We observed consistent outlier across various model chemistries in vibrational frequency predictions, with alternative scaling factors showing minimal impact on reducing outlier occurrences. Besides, large basis sets are not indispensably required for fundamental frequency predictions. M06-L, revM06-L, SOGGA11-X, PW6B95-D3(BJ), CF22D, and M06-2X consistently exhibit excellent performance across the three basis sets. When using frequency-range-specific scaling factors, the root mean squard errors (RMSEs) and median absolute errors (MedAEs) of almost all the selected functionals were reduced. PW6B95-D3(BJ), CF22D, and MN12-SX exhibited the lowest RMSEs. Comparisons were also done for different data classifications; the dataset was classified by the elements of the molecules, vibrational frequency intervals, and vibrational modes.</p>\n </div>","PeriodicalId":182,"journal":{"name":"International Journal of Quantum Chemistry","volume":"124 22","pages":""},"PeriodicalIF":2.7000,"publicationDate":"2024-11-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Performance of Minnesota Functionals on Vibrational Frequency\",\"authors\":\"Jiaxu Wang, Cheng Zhang, Yaqi Li, Yini Zhou, Yuanyuan Shu, Songping Liang, Gaihua Zhang, Zhonghua Liu, Ying Wang\",\"doi\":\"10.1002/qua.27516\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n <p>Molecular geometry and harmonic frequency calculations are essential in thermochemical computations, with density functional theory (DFT) being widely employed for vibrational frequency predictions due to its efficiency and accuracy. In this study, we assessed the precision of 28 Minnesota based functionals with three different basis sets using the VIBFREQ1295 dataset. Scaling factors are necessary for predicting fundamental frequencies, global scaling factors were fitted by using F38/10 and VIBFREQ1295 datasets. The superior performing functionals were then fitted based on vibrational frequency ranges to obtain frequency-range-specific scaling factors. We observed consistent outlier across various model chemistries in vibrational frequency predictions, with alternative scaling factors showing minimal impact on reducing outlier occurrences. Besides, large basis sets are not indispensably required for fundamental frequency predictions. M06-L, revM06-L, SOGGA11-X, PW6B95-D3(BJ), CF22D, and M06-2X consistently exhibit excellent performance across the three basis sets. When using frequency-range-specific scaling factors, the root mean squard errors (RMSEs) and median absolute errors (MedAEs) of almost all the selected functionals were reduced. PW6B95-D3(BJ), CF22D, and MN12-SX exhibited the lowest RMSEs. Comparisons were also done for different data classifications; the dataset was classified by the elements of the molecules, vibrational frequency intervals, and vibrational modes.</p>\\n </div>\",\"PeriodicalId\":182,\"journal\":{\"name\":\"International Journal of Quantum Chemistry\",\"volume\":\"124 22\",\"pages\":\"\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2024-11-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Quantum Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/qua.27516\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Quantum Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/qua.27516","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Performance of Minnesota Functionals on Vibrational Frequency
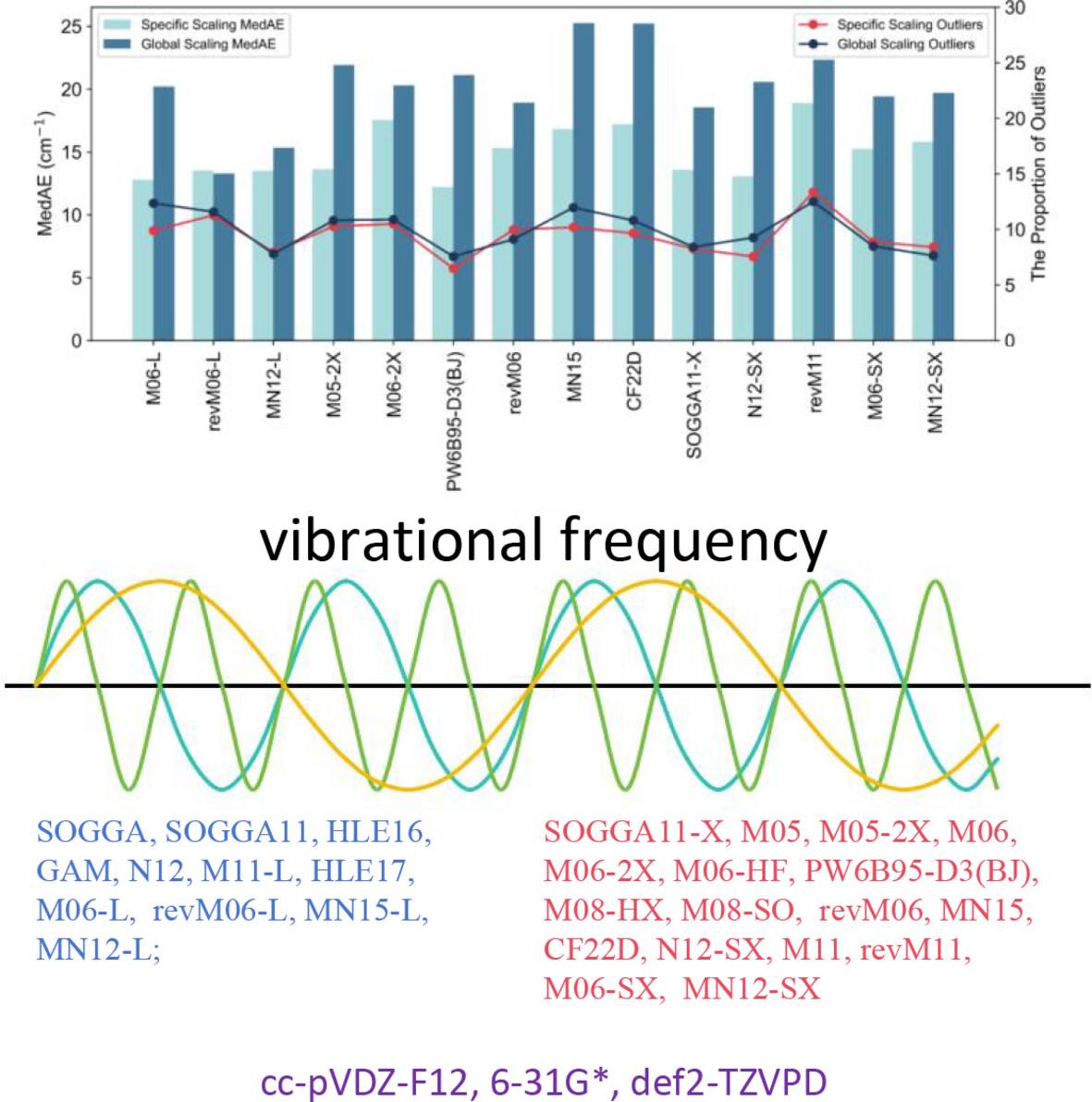
Molecular geometry and harmonic frequency calculations are essential in thermochemical computations, with density functional theory (DFT) being widely employed for vibrational frequency predictions due to its efficiency and accuracy. In this study, we assessed the precision of 28 Minnesota based functionals with three different basis sets using the VIBFREQ1295 dataset. Scaling factors are necessary for predicting fundamental frequencies, global scaling factors were fitted by using F38/10 and VIBFREQ1295 datasets. The superior performing functionals were then fitted based on vibrational frequency ranges to obtain frequency-range-specific scaling factors. We observed consistent outlier across various model chemistries in vibrational frequency predictions, with alternative scaling factors showing minimal impact on reducing outlier occurrences. Besides, large basis sets are not indispensably required for fundamental frequency predictions. M06-L, revM06-L, SOGGA11-X, PW6B95-D3(BJ), CF22D, and M06-2X consistently exhibit excellent performance across the three basis sets. When using frequency-range-specific scaling factors, the root mean squard errors (RMSEs) and median absolute errors (MedAEs) of almost all the selected functionals were reduced. PW6B95-D3(BJ), CF22D, and MN12-SX exhibited the lowest RMSEs. Comparisons were also done for different data classifications; the dataset was classified by the elements of the molecules, vibrational frequency intervals, and vibrational modes.
期刊介绍:
Since its first formulation quantum chemistry has provided the conceptual and terminological framework necessary to understand atoms, molecules and the condensed matter. Over the past decades synergistic advances in the methodological developments, software and hardware have transformed quantum chemistry in a truly interdisciplinary science that has expanded beyond its traditional core of molecular sciences to fields as diverse as chemistry and catalysis, biophysics, nanotechnology and material science.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
