Igor V. Polyakov , Kirill D. Miroshnichenko , Tatiana I. Mulashkina , Alexander A. Moskovsky , Ekaterina I. Marchenko , Maria G. Khrenova
{"title":"迈向准确的机器学习预测ATP酶解时P-O键切割的性质","authors":"Igor V. Polyakov , Kirill D. Miroshnichenko , Tatiana I. Mulashkina , Alexander A. Moskovsky , Ekaterina I. Marchenko , Maria G. Khrenova","doi":"10.1016/j.mencom.2024.10.003","DOIUrl":null,"url":null,"abstract":"<div><div>Molecular dynamic simulations using QM/MM potentials are performed for the enzyme–substrate complex of adenosine triphosphate (ATP) with the motor protein myosin. Machine learning methods are applied to a dataset consisting of the geometry parameters of the active site in the enzyme–substrate complex to predict the Laplacian of electron density at the bond critical point of the P<sub>G</sub>–O<sub>3B</sub> bond being broken in ATP. Using a gradient boosting machine learning model, a mean absolute error of 0.01 a.u. and an <em>R</em><sup>2</sup> score of 0.99 are achieved, and it is found that the P<sub>G</sub>–O<sub>3B</sub> bond length is the most important feature, contributing 2/3, while other geometry features contribute 1/3.</div></div>","PeriodicalId":18542,"journal":{"name":"Mendeleev Communications","volume":"34 6","pages":"Pages 776-779"},"PeriodicalIF":1.7000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Towards accurate machine learning predictions of properties of the P–O bond cleaving in ATP upon enzymatic hydrolysis\",\"authors\":\"Igor V. Polyakov , Kirill D. Miroshnichenko , Tatiana I. Mulashkina , Alexander A. Moskovsky , Ekaterina I. Marchenko , Maria G. Khrenova\",\"doi\":\"10.1016/j.mencom.2024.10.003\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Molecular dynamic simulations using QM/MM potentials are performed for the enzyme–substrate complex of adenosine triphosphate (ATP) with the motor protein myosin. Machine learning methods are applied to a dataset consisting of the geometry parameters of the active site in the enzyme–substrate complex to predict the Laplacian of electron density at the bond critical point of the P<sub>G</sub>–O<sub>3B</sub> bond being broken in ATP. Using a gradient boosting machine learning model, a mean absolute error of 0.01 a.u. and an <em>R</em><sup>2</sup> score of 0.99 are achieved, and it is found that the P<sub>G</sub>–O<sub>3B</sub> bond length is the most important feature, contributing 2/3, while other geometry features contribute 1/3.</div></div>\",\"PeriodicalId\":18542,\"journal\":{\"name\":\"Mendeleev Communications\",\"volume\":\"34 6\",\"pages\":\"Pages 776-779\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Mendeleev Communications\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0959943624003018\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/29 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Mendeleev Communications","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0959943624003018","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/29 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
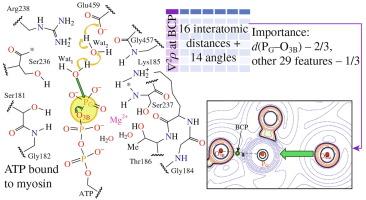
Towards accurate machine learning predictions of properties of the P–O bond cleaving in ATP upon enzymatic hydrolysis
Molecular dynamic simulations using QM/MM potentials are performed for the enzyme–substrate complex of adenosine triphosphate (ATP) with the motor protein myosin. Machine learning methods are applied to a dataset consisting of the geometry parameters of the active site in the enzyme–substrate complex to predict the Laplacian of electron density at the bond critical point of the PG–O3B bond being broken in ATP. Using a gradient boosting machine learning model, a mean absolute error of 0.01 a.u. and an R2 score of 0.99 are achieved, and it is found that the PG–O3B bond length is the most important feature, contributing 2/3, while other geometry features contribute 1/3.
期刊介绍:
Mendeleev Communications is the journal of the Russian Academy of Sciences, launched jointly by the Academy of Sciences of the USSR and the Royal Society of Chemistry (United Kingdom) in 1991. Starting from 1st January 2007, Elsevier is the new publishing partner of Mendeleev Communications.
Mendeleev Communications publishes short communications in chemistry. The journal primarily features papers from the Russian Federation and the other states of the former USSR. However, it also includes papers by authors from other parts of the world. Mendeleev Communications is not a translated journal, but instead is published directly in English. The International Editorial Board is composed of eminent scientists who provide advice on refereeing policy.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
