Brooke P Quertermous, Hayley J Hawkins, Alyssa A Schlotman, Huiying Wang, Sarah Kemme, Anita Pai, Settapong Jitwongwai, Napat Angkathunyakul, Ananya Pongpaibul, Saeed Mohammad
{"title":"一种新的遗传变异与进行性家族性肝内胆汁淤积3型相关:一个病例系列。","authors":"Brooke P Quertermous, Hayley J Hawkins, Alyssa A Schlotman, Huiying Wang, Sarah Kemme, Anita Pai, Settapong Jitwongwai, Napat Angkathunyakul, Ananya Pongpaibul, Saeed Mohammad","doi":"10.1002/jpr3.12119","DOIUrl":null,"url":null,"abstract":"<p><p>Progressive familial intrahepatic cholestasis type 3 (PFIC-3) is a rare disorder characterized by chronic cholestasis usually progressing to end-stage liver disease (ESLD) within the first two decades of life. PFIC-3 is caused by pathogenic genetic variants of the ATP-binding cassette 4 (ABCB4) gene with variable inheritance; the most common is autosomal recessive. We present two cases of PFIC-3 with genetic testing confirming a novel genetic variant in ABCB4 with homozygous genotype c.779 T > C, p.L260P. Both individuals are from mainland Southeast Asia and have a clinical picture consistent with cholestasis progressing to ESLD.</p>","PeriodicalId":501015,"journal":{"name":"JPGN reports","volume":"5 4","pages":"538-541"},"PeriodicalIF":0.0000,"publicationDate":"2024-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11600367/pdf/","citationCount":"0","resultStr":"{\"title\":\"A novel genetic variant associated with progressive familial intrahepatic cholestasis type 3: A case series.\",\"authors\":\"Brooke P Quertermous, Hayley J Hawkins, Alyssa A Schlotman, Huiying Wang, Sarah Kemme, Anita Pai, Settapong Jitwongwai, Napat Angkathunyakul, Ananya Pongpaibul, Saeed Mohammad\",\"doi\":\"10.1002/jpr3.12119\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Progressive familial intrahepatic cholestasis type 3 (PFIC-3) is a rare disorder characterized by chronic cholestasis usually progressing to end-stage liver disease (ESLD) within the first two decades of life. PFIC-3 is caused by pathogenic genetic variants of the ATP-binding cassette 4 (ABCB4) gene with variable inheritance; the most common is autosomal recessive. We present two cases of PFIC-3 with genetic testing confirming a novel genetic variant in ABCB4 with homozygous genotype c.779 T > C, p.L260P. Both individuals are from mainland Southeast Asia and have a clinical picture consistent with cholestasis progressing to ESLD.</p>\",\"PeriodicalId\":501015,\"journal\":{\"name\":\"JPGN reports\",\"volume\":\"5 4\",\"pages\":\"538-541\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-09-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11600367/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JPGN reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1002/jpr3.12119\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JPGN reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1002/jpr3.12119","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
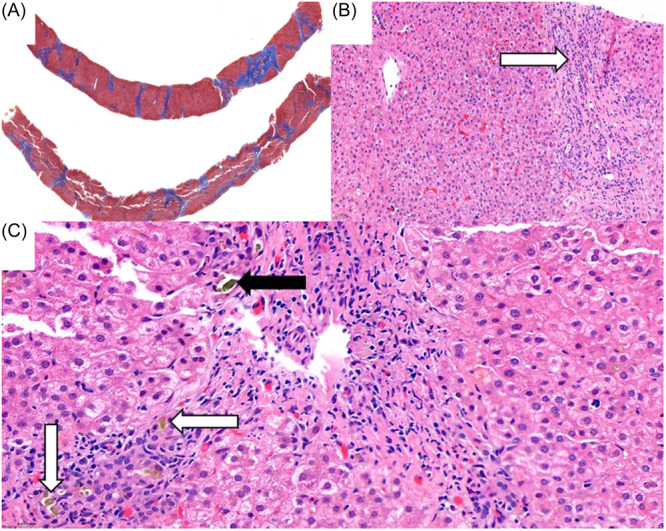
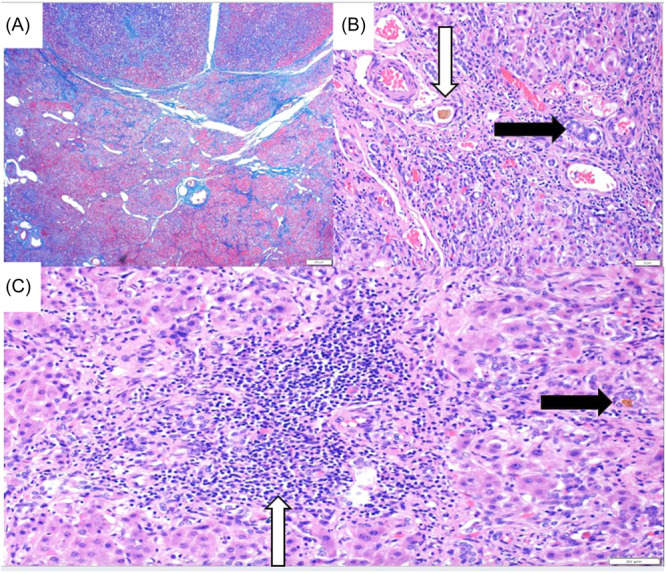
进行性家族性肝内胆汁淤积3型(PFIC-3)是一种罕见的以慢性胆汁淤积为特征的疾病,通常在生命的前20年进展为终末期肝病(ESLD)。PFIC-3是由atp结合盒4 (ABCB4)基因的致病性遗传变异引起的,具有可变遗传;最常见的是常染色体隐性遗传。我们报告了两例PFIC-3的基因检测,证实了ABCB4纯合子基因型C .779 T . > C . p.L260P的新遗传变异。这两名患者均来自东南亚大陆,其临床表现与胆汁淤积进展为ESLD一致。
A novel genetic variant associated with progressive familial intrahepatic cholestasis type 3: A case series.
Progressive familial intrahepatic cholestasis type 3 (PFIC-3) is a rare disorder characterized by chronic cholestasis usually progressing to end-stage liver disease (ESLD) within the first two decades of life. PFIC-3 is caused by pathogenic genetic variants of the ATP-binding cassette 4 (ABCB4) gene with variable inheritance; the most common is autosomal recessive. We present two cases of PFIC-3 with genetic testing confirming a novel genetic variant in ABCB4 with homozygous genotype c.779 T > C, p.L260P. Both individuals are from mainland Southeast Asia and have a clinical picture consistent with cholestasis progressing to ESLD.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
