Lara Fuhrmann, Benjamin Langer, Ivan Topolsky, Niko Beerenwinkel
{"title":"VILOCA:测序质量敏感的病毒单倍型重建和突变,需要短读和长读数据。","authors":"Lara Fuhrmann, Benjamin Langer, Ivan Topolsky, Niko Beerenwinkel","doi":"10.1093/nargab/lqae152","DOIUrl":null,"url":null,"abstract":"<p><p>RNA viruses exist as large heterogeneous populations within their host. The structure and diversity of virus populations affects disease progression and treatment outcomes. Next-generation sequencing allows detailed viral population analysis, but inferring diversity from error-prone reads is challenging. Here, we present VILOCA (VIral LOcal haplotype reconstruction and mutation CAlling for short and long read data), a method for mutation calling and reconstruction of local haplotypes from short- and long-read viral sequencing data. Local haplotypes refer to genomic regions that have approximately the length of the input reads. VILOCA recovers local haplotypes by using a Dirichlet process mixture model to cluster reads around their unobserved haplotypes and leveraging quality scores of the sequencing reads. We assessed the performance of VILOCA in terms of mutation calling and haplotype reconstruction accuracy on simulated and experimental Illumina, PacBio and Oxford Nanopore data. On simulated and experimental Illumina data, VILOCA performed better or similar to existing methods. On the simulated long-read data, VILOCA is able to recover on average [Formula: see text] of the ground truth mutations with perfect precision compared to only [Formula: see text] recall and [Formula: see text] precision of the second-best method. In summary, VILOCA provides significantly improved accuracy in mutation and haplotype calling, especially for long-read sequencing data, and therefore facilitates the comprehensive characterization of heterogeneous within-host viral populations.</p>","PeriodicalId":33994,"journal":{"name":"NAR Genomics and Bioinformatics","volume":"6 4","pages":"lqae152"},"PeriodicalIF":3.0000,"publicationDate":"2024-11-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11616694/pdf/","citationCount":"0","resultStr":"{\"title\":\"VILOCA: sequencing quality-aware viral haplotype reconstruction and mutation calling for short-read and long-read data.\",\"authors\":\"Lara Fuhrmann, Benjamin Langer, Ivan Topolsky, Niko Beerenwinkel\",\"doi\":\"10.1093/nargab/lqae152\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>RNA viruses exist as large heterogeneous populations within their host. The structure and diversity of virus populations affects disease progression and treatment outcomes. Next-generation sequencing allows detailed viral population analysis, but inferring diversity from error-prone reads is challenging. Here, we present VILOCA (VIral LOcal haplotype reconstruction and mutation CAlling for short and long read data), a method for mutation calling and reconstruction of local haplotypes from short- and long-read viral sequencing data. Local haplotypes refer to genomic regions that have approximately the length of the input reads. VILOCA recovers local haplotypes by using a Dirichlet process mixture model to cluster reads around their unobserved haplotypes and leveraging quality scores of the sequencing reads. We assessed the performance of VILOCA in terms of mutation calling and haplotype reconstruction accuracy on simulated and experimental Illumina, PacBio and Oxford Nanopore data. On simulated and experimental Illumina data, VILOCA performed better or similar to existing methods. On the simulated long-read data, VILOCA is able to recover on average [Formula: see text] of the ground truth mutations with perfect precision compared to only [Formula: see text] recall and [Formula: see text] precision of the second-best method. In summary, VILOCA provides significantly improved accuracy in mutation and haplotype calling, especially for long-read sequencing data, and therefore facilitates the comprehensive characterization of heterogeneous within-host viral populations.</p>\",\"PeriodicalId\":33994,\"journal\":{\"name\":\"NAR Genomics and Bioinformatics\",\"volume\":\"6 4\",\"pages\":\"lqae152\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-11-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11616694/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NAR Genomics and Bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/nargab/lqae152\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/12/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Genomics and Bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/nargab/lqae152","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/12/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
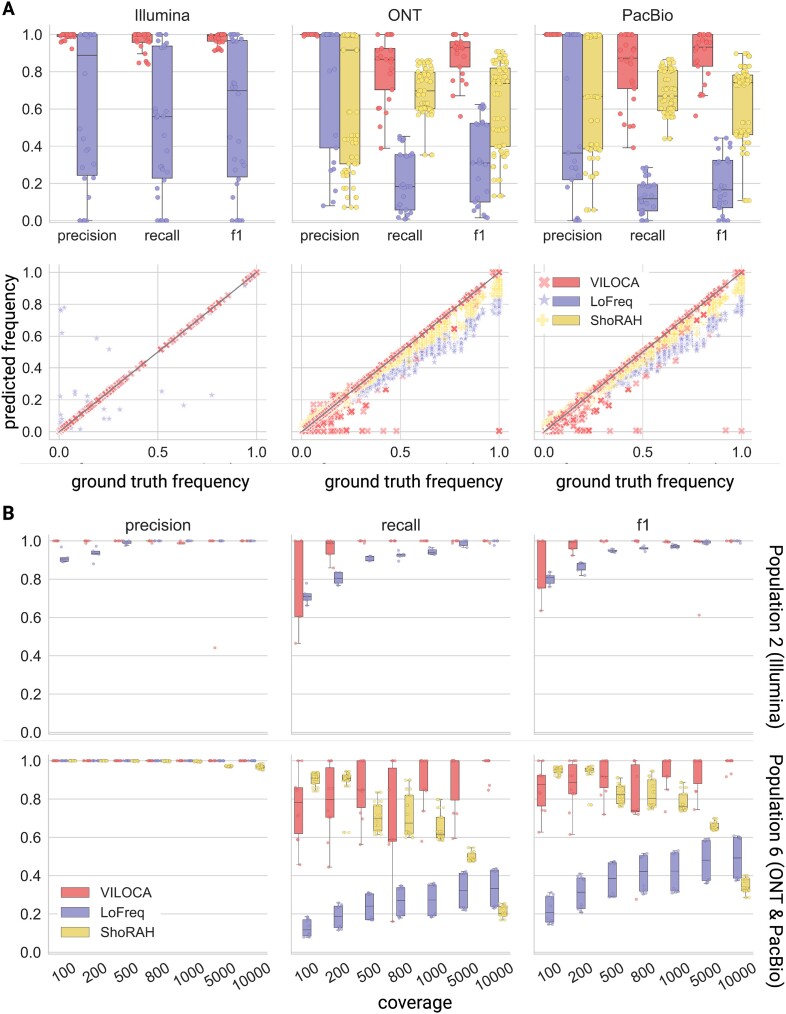
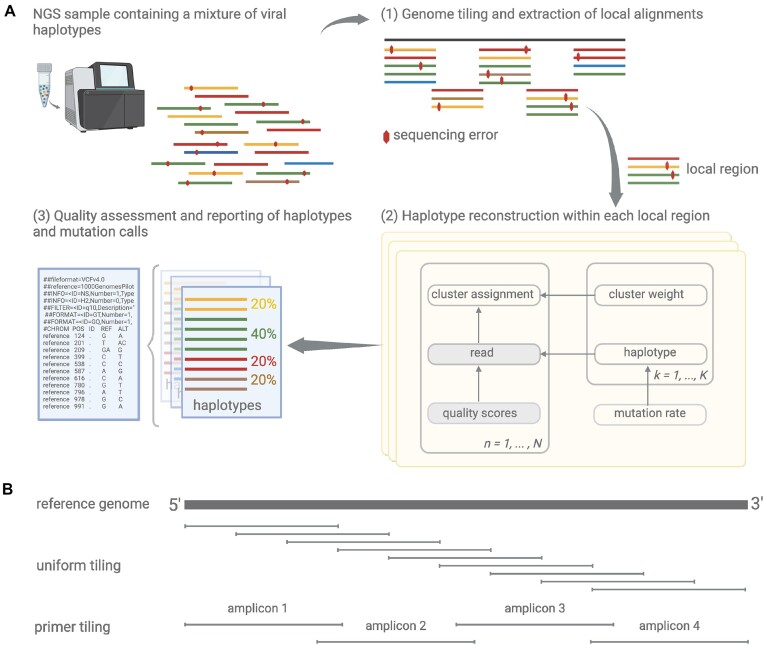
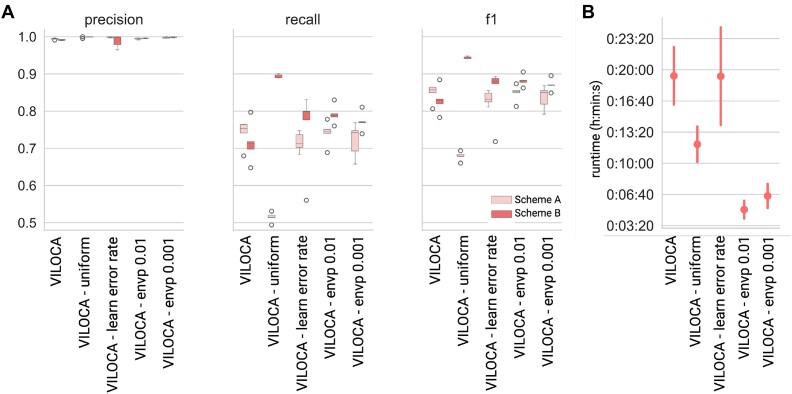
RNA病毒在宿主体内以大量异质群体的形式存在。病毒种群的结构和多样性影响疾病进展和治疗结果。下一代测序允许详细的病毒种群分析,但从容易出错的读取推断多样性是具有挑战性的。在这里,我们提出了VILOCA (VIral LOcal haplotype reconstruction and mutation CAlling for short and long read data),这是一种从短读和长读病毒测序数据中调用突变和重建局部单倍型的方法。局部单倍型指的是基因组区域,其长度与输入序列的长度大致相同。VILOCA通过使用Dirichlet过程混合模型在未观察到的单倍型周围聚类读取并利用测序读取的质量分数来恢复局部单倍型。我们在模拟和实验Illumina、PacBio和Oxford Nanopore数据上评估了VILOCA在突变召唤和单倍型重建精度方面的表现。在模拟和实验Illumina数据上,VILOCA的表现比现有方法更好或相似。在模拟的长读数据上,VILOCA能够以完美的精度平均恢复[公式:参见文本]的ground truth突变,而只有[公式:参见文本]recall和[公式:参见文本]precision的次优方法。总之,VILOCA显著提高了突变和单倍型召唤的准确性,特别是对于长读测序数据,因此有助于全面表征宿主病毒群体内的异质性。
VILOCA: sequencing quality-aware viral haplotype reconstruction and mutation calling for short-read and long-read data.
RNA viruses exist as large heterogeneous populations within their host. The structure and diversity of virus populations affects disease progression and treatment outcomes. Next-generation sequencing allows detailed viral population analysis, but inferring diversity from error-prone reads is challenging. Here, we present VILOCA (VIral LOcal haplotype reconstruction and mutation CAlling for short and long read data), a method for mutation calling and reconstruction of local haplotypes from short- and long-read viral sequencing data. Local haplotypes refer to genomic regions that have approximately the length of the input reads. VILOCA recovers local haplotypes by using a Dirichlet process mixture model to cluster reads around their unobserved haplotypes and leveraging quality scores of the sequencing reads. We assessed the performance of VILOCA in terms of mutation calling and haplotype reconstruction accuracy on simulated and experimental Illumina, PacBio and Oxford Nanopore data. On simulated and experimental Illumina data, VILOCA performed better or similar to existing methods. On the simulated long-read data, VILOCA is able to recover on average [Formula: see text] of the ground truth mutations with perfect precision compared to only [Formula: see text] recall and [Formula: see text] precision of the second-best method. In summary, VILOCA provides significantly improved accuracy in mutation and haplotype calling, especially for long-read sequencing data, and therefore facilitates the comprehensive characterization of heterogeneous within-host viral populations.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
