{"title":"通过整合大规模转录组数据集,植物表型预测得到了改善。","authors":"Zefeng Wu, Yali Sun, Xiaoqiang Zhao, Zigang Liu, Wenqi Zhou, Yining Niu","doi":"10.1093/nargab/lqae184","DOIUrl":null,"url":null,"abstract":"<p><p>Research on the dynamic expression of genes in plants is important for understanding different biological processes. We used the large amounts of transcriptomic data from various plant sample sources that are publicly available to investigate whether the expression levels of a subset of highly variable genes (HVGs) can be used to accurately identify the phenotypes of plants. Using maize (<i>Zea mays</i> L.) as an example, we built machine learning (ML) models to predict phenotypes using a gene expression dataset of 21 612 bulk RNA sequencing samples. We showed that the ML models achieved excellent prediction accuracy using only the HVGs to identify different phenotypes, including tissue types, developmental stages, cultivars and stress conditions. By ML models, several important functional genes were found to be associated with different phenotypes. We performed a similar analysis in rice (<i>Orzya sativa</i> L.) and found that the ML models could be generalized across species. However, the models trained from maize did not perform well in rice, probably because of the expression divergence of the conserved HVGs between the two species. Overall, our results provide an ML framework for phenotype prediction using gene expression profiles, which may contribute to precision management of crops in agricultural practices.</p>","PeriodicalId":33994,"journal":{"name":"NAR Genomics and Bioinformatics","volume":"6 4","pages":"lqae184"},"PeriodicalIF":2.8000,"publicationDate":"2024-12-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11672113/pdf/","citationCount":"0","resultStr":"{\"title\":\"Phenotype prediction in plants is improved by integrating large-scale transcriptomic datasets.\",\"authors\":\"Zefeng Wu, Yali Sun, Xiaoqiang Zhao, Zigang Liu, Wenqi Zhou, Yining Niu\",\"doi\":\"10.1093/nargab/lqae184\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Research on the dynamic expression of genes in plants is important for understanding different biological processes. We used the large amounts of transcriptomic data from various plant sample sources that are publicly available to investigate whether the expression levels of a subset of highly variable genes (HVGs) can be used to accurately identify the phenotypes of plants. Using maize (<i>Zea mays</i> L.) as an example, we built machine learning (ML) models to predict phenotypes using a gene expression dataset of 21 612 bulk RNA sequencing samples. We showed that the ML models achieved excellent prediction accuracy using only the HVGs to identify different phenotypes, including tissue types, developmental stages, cultivars and stress conditions. By ML models, several important functional genes were found to be associated with different phenotypes. We performed a similar analysis in rice (<i>Orzya sativa</i> L.) and found that the ML models could be generalized across species. However, the models trained from maize did not perform well in rice, probably because of the expression divergence of the conserved HVGs between the two species. Overall, our results provide an ML framework for phenotype prediction using gene expression profiles, which may contribute to precision management of crops in agricultural practices.</p>\",\"PeriodicalId\":33994,\"journal\":{\"name\":\"NAR Genomics and Bioinformatics\",\"volume\":\"6 4\",\"pages\":\"lqae184\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-12-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11672113/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NAR Genomics and Bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/nargab/lqae184\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/12/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Genomics and Bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/nargab/lqae184","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/12/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Phenotype prediction in plants is improved by integrating large-scale transcriptomic datasets.
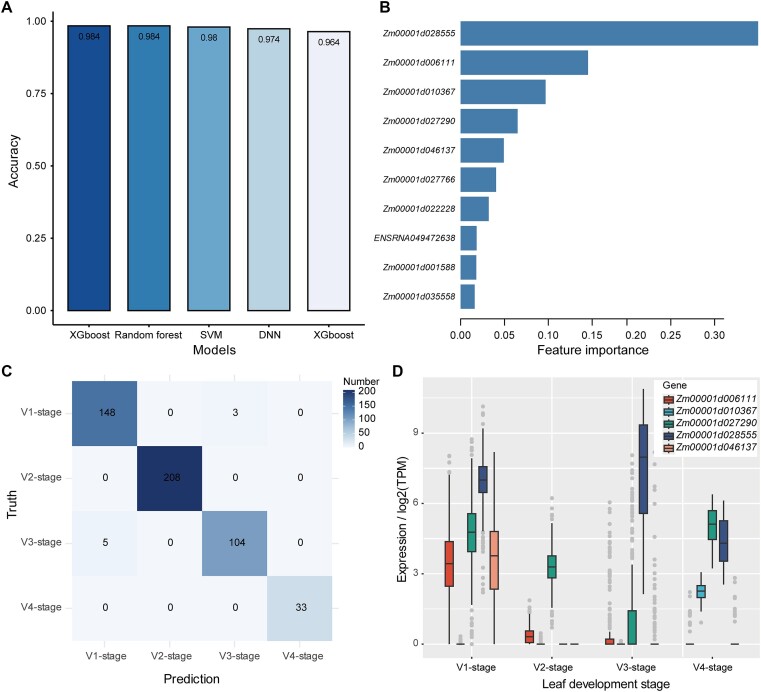
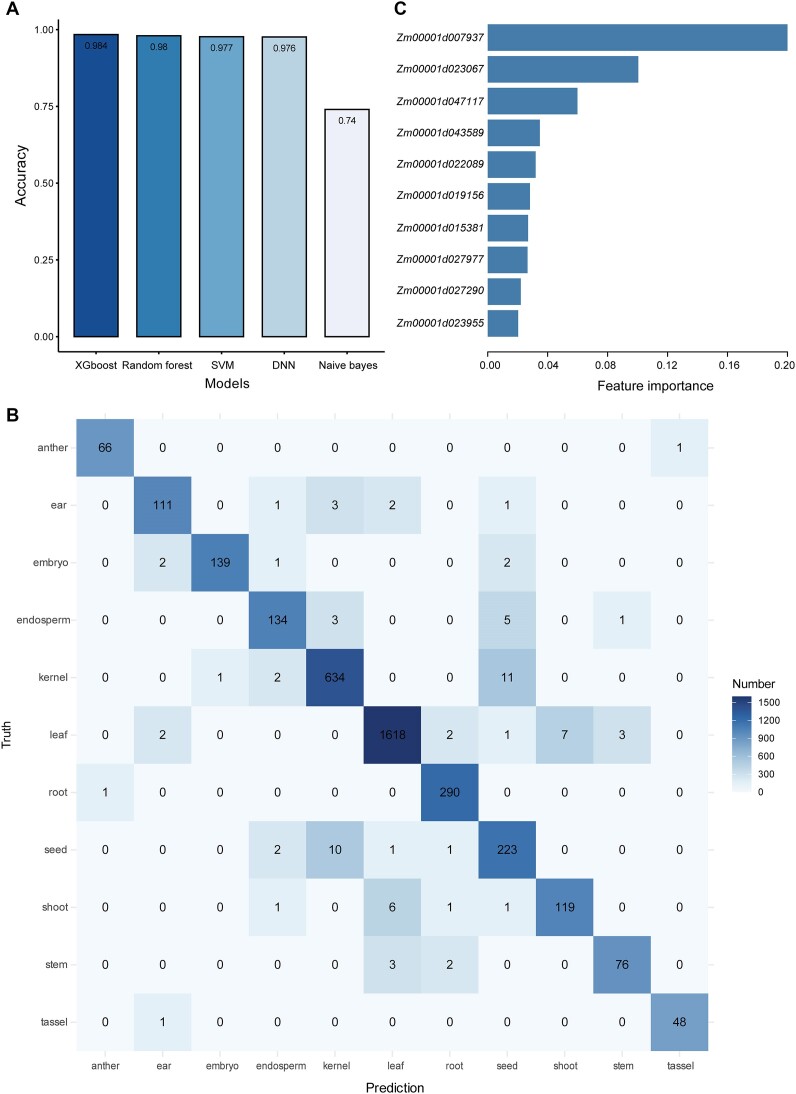
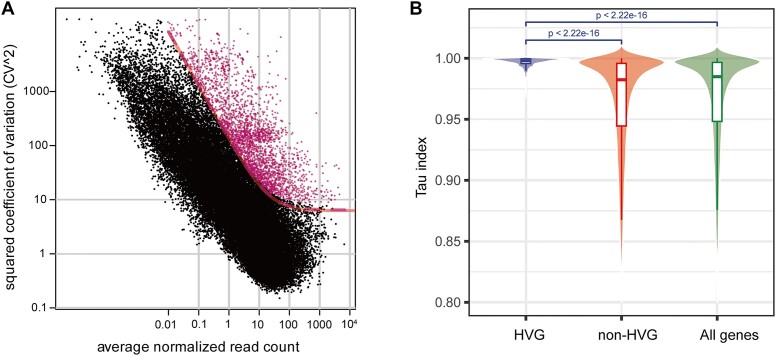
Research on the dynamic expression of genes in plants is important for understanding different biological processes. We used the large amounts of transcriptomic data from various plant sample sources that are publicly available to investigate whether the expression levels of a subset of highly variable genes (HVGs) can be used to accurately identify the phenotypes of plants. Using maize (Zea mays L.) as an example, we built machine learning (ML) models to predict phenotypes using a gene expression dataset of 21 612 bulk RNA sequencing samples. We showed that the ML models achieved excellent prediction accuracy using only the HVGs to identify different phenotypes, including tissue types, developmental stages, cultivars and stress conditions. By ML models, several important functional genes were found to be associated with different phenotypes. We performed a similar analysis in rice (Orzya sativa L.) and found that the ML models could be generalized across species. However, the models trained from maize did not perform well in rice, probably because of the expression divergence of the conserved HVGs between the two species. Overall, our results provide an ML framework for phenotype prediction using gene expression profiles, which may contribute to precision management of crops in agricultural practices.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
