Andrea Chen, Shannon Louise Hart, Melissa Lannon, Cynthia Hawkins, Kesava K V Reddy, Jian-Qiang Lu
{"title":"鲁宾斯坦-泰比综合征中的脑膜瘤:病例报告和综合综述。","authors":"Andrea Chen, Shannon Louise Hart, Melissa Lannon, Cynthia Hawkins, Kesava K V Reddy, Jian-Qiang Lu","doi":"10.1093/jnen/nlae135","DOIUrl":null,"url":null,"abstract":"<p><p>Rubinstein-Taybi syndrome (RTS) is a congenital disorder with characteristic clinical manifestations. In the vast majority of cases, it is caused by mutations of the gene encoding the transcriptional co-activator cAMP-response element binding protein (CBP)-binding protein (CREBBP). It has been thought to be a tumor predisposition syndrome as RTS patients have an increased risk of developing tumors including meningiomas. However, RTS-associated meningiomas are rarely reported. We report a unique RTS-associated meningioma in which an oncogenic CREBBP mutation is identified. We also comprehensively review the reported RTS-associated meningiomas, from epidemiology and pathogenesis to clinicopathological characteristics and treatment. All RTS patients with meningiomas are female and have the exclusive mutations of CREBBP. In population-based studies RTS-associated meningiomas seem to develop at younger ages. Their pathogenesis may be driven by the CREBBP/CBP alterations resulting in aberrant signal transduction in the CBP-mediated signaling pathways. Meningiomas in RTS patients have common clinicopathological characteristics including comorbidity with other tumors, radiologically intra-osseous growth, and uncommon histopathology such as ossifying and secretory features. Given the genetic nature and rarity of RTS-associated meningiomas, further investigation of their characteristics may define molecular targets for improved therapeutic options for RTS patients.</p>","PeriodicalId":16682,"journal":{"name":"Journal of Neuropathology and Experimental Neurology","volume":" ","pages":"329-336"},"PeriodicalIF":3.0000,"publicationDate":"2025-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11923739/pdf/","citationCount":"0","resultStr":"{\"title\":\"Meningiomas in Rubinstein-Taybi syndrome: A case report and comprehensive review.\",\"authors\":\"Andrea Chen, Shannon Louise Hart, Melissa Lannon, Cynthia Hawkins, Kesava K V Reddy, Jian-Qiang Lu\",\"doi\":\"10.1093/jnen/nlae135\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Rubinstein-Taybi syndrome (RTS) is a congenital disorder with characteristic clinical manifestations. In the vast majority of cases, it is caused by mutations of the gene encoding the transcriptional co-activator cAMP-response element binding protein (CBP)-binding protein (CREBBP). It has been thought to be a tumor predisposition syndrome as RTS patients have an increased risk of developing tumors including meningiomas. However, RTS-associated meningiomas are rarely reported. We report a unique RTS-associated meningioma in which an oncogenic CREBBP mutation is identified. We also comprehensively review the reported RTS-associated meningiomas, from epidemiology and pathogenesis to clinicopathological characteristics and treatment. All RTS patients with meningiomas are female and have the exclusive mutations of CREBBP. In population-based studies RTS-associated meningiomas seem to develop at younger ages. Their pathogenesis may be driven by the CREBBP/CBP alterations resulting in aberrant signal transduction in the CBP-mediated signaling pathways. Meningiomas in RTS patients have common clinicopathological characteristics including comorbidity with other tumors, radiologically intra-osseous growth, and uncommon histopathology such as ossifying and secretory features. Given the genetic nature and rarity of RTS-associated meningiomas, further investigation of their characteristics may define molecular targets for improved therapeutic options for RTS patients.</p>\",\"PeriodicalId\":16682,\"journal\":{\"name\":\"Journal of Neuropathology and Experimental Neurology\",\"volume\":\" \",\"pages\":\"329-336\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2025-04-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11923739/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Neuropathology and Experimental Neurology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1093/jnen/nlae135\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Neuropathology and Experimental Neurology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1093/jnen/nlae135","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
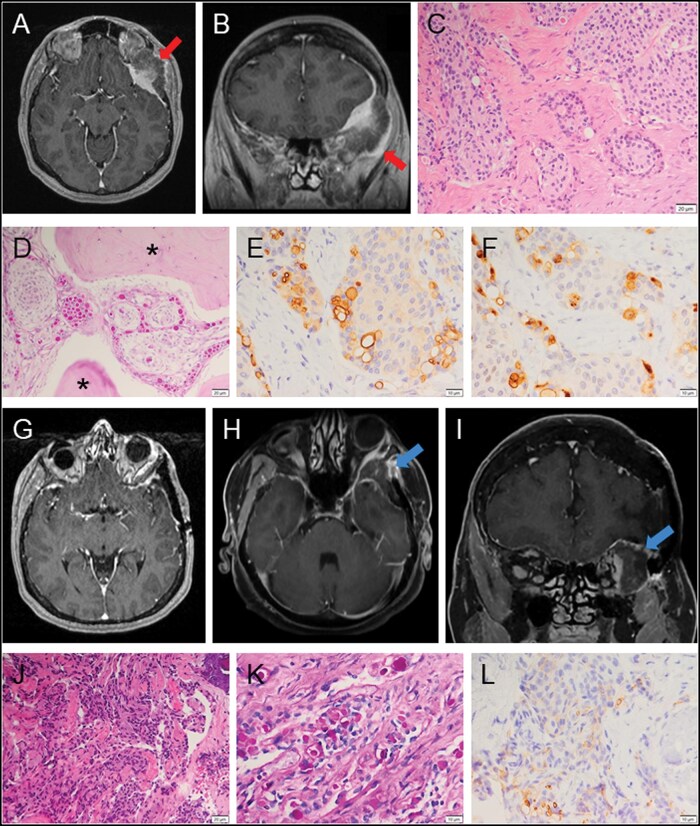
Meningiomas in Rubinstein-Taybi syndrome: A case report and comprehensive review.
Rubinstein-Taybi syndrome (RTS) is a congenital disorder with characteristic clinical manifestations. In the vast majority of cases, it is caused by mutations of the gene encoding the transcriptional co-activator cAMP-response element binding protein (CBP)-binding protein (CREBBP). It has been thought to be a tumor predisposition syndrome as RTS patients have an increased risk of developing tumors including meningiomas. However, RTS-associated meningiomas are rarely reported. We report a unique RTS-associated meningioma in which an oncogenic CREBBP mutation is identified. We also comprehensively review the reported RTS-associated meningiomas, from epidemiology and pathogenesis to clinicopathological characteristics and treatment. All RTS patients with meningiomas are female and have the exclusive mutations of CREBBP. In population-based studies RTS-associated meningiomas seem to develop at younger ages. Their pathogenesis may be driven by the CREBBP/CBP alterations resulting in aberrant signal transduction in the CBP-mediated signaling pathways. Meningiomas in RTS patients have common clinicopathological characteristics including comorbidity with other tumors, radiologically intra-osseous growth, and uncommon histopathology such as ossifying and secretory features. Given the genetic nature and rarity of RTS-associated meningiomas, further investigation of their characteristics may define molecular targets for improved therapeutic options for RTS patients.
期刊介绍:
Journal of Neuropathology & Experimental Neurology is the official journal of the American Association of Neuropathologists, Inc. (AANP). The journal publishes peer-reviewed studies on neuropathology and experimental neuroscience, book reviews, letters, and Association news, covering a broad spectrum of fields in basic neuroscience with an emphasis on human neurological diseases. It is written by and for neuropathologists, neurologists, neurosurgeons, pathologists, psychiatrists, and basic neuroscientists from around the world. Publication has been continuous since 1942.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
