Lin Yue , Zui Tan , Wei Wei , Hongyao Liu , Taixiong Xue , Xingping Su , Xiuli Wu , Yuting Xie , Peilin Li , Doudou Wang , Zhihao Liu , Cailing Gan , Tinghong Ye
{"title":"一种有效的口服FGFRs抑制剂的设计、合成和生物学评价","authors":"Lin Yue , Zui Tan , Wei Wei , Hongyao Liu , Taixiong Xue , Xingping Su , Xiuli Wu , Yuting Xie , Peilin Li , Doudou Wang , Zhihao Liu , Cailing Gan , Tinghong Ye","doi":"10.1016/j.ejmech.2024.117232","DOIUrl":null,"url":null,"abstract":"<div><div>Organ fibrosis, such as lung fibrosis and liver fibrosis, is a progressive and fatal disease. Fibroblast growth factor receptors (FGFRs) play an important role in the development and progression of fibrosis. Through scaffold hopping, bioisosteric replacement design, and structure-activity relationship optimization, we developed a series of highly potent FGFRs inhibitors, and the indazole-containing candidate compound <strong>A16</strong> showed potent kinase activity comparable to that of <strong>AZD4547</strong>. In addition, <strong>A16</strong> effectively suppressed the activation of lung fibroblasts and hepatic stellate cells (HSCs) induced by TGF-β1, leading to a reduction in collagen deposition. Notably, <strong>A16</strong> exhibited potent anti-fibrotic effects through the inhibition of the FGFR pathway <em>in vitro</em>. Compound <strong>A16</strong> also showed reasonable pharmacokinetic properties (<em>F</em> = 21.84 %) and favorable cardiac safety (hERG IC<sub>50</sub> > 20 μM). Moreover, in models of pulmonary fibrosis, <strong>A16</strong> ameliorated (in the prevention model) and reversed (in the treatment model) bleomycin-induced lung fibrosis, as well as mitigated inflammatory immune response in the lung. Furthermore, in the CCl<sub>4</sub>-induced liver fibrosis model, when <strong>A16</strong> was administrated orally at a dose of 30 mg/kg/day for 3 weeks, it effectively improved liver function, restored damaged liver structures, and reduced collagen deposition. Taken together, these results suggest that <strong>A16</strong> could be a potential drug candidate for the treatment of organ fibrosis.</div></div>","PeriodicalId":314,"journal":{"name":"European Journal of Medicinal Chemistry","volume":"285 ","pages":"Article 117232"},"PeriodicalIF":5.9000,"publicationDate":"2025-03-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Design, synthesis, and biological evaluation of a potent and orally bioavailable FGFRs inhibitor for fibrotic treatment\",\"authors\":\"Lin Yue , Zui Tan , Wei Wei , Hongyao Liu , Taixiong Xue , Xingping Su , Xiuli Wu , Yuting Xie , Peilin Li , Doudou Wang , Zhihao Liu , Cailing Gan , Tinghong Ye\",\"doi\":\"10.1016/j.ejmech.2024.117232\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Organ fibrosis, such as lung fibrosis and liver fibrosis, is a progressive and fatal disease. Fibroblast growth factor receptors (FGFRs) play an important role in the development and progression of fibrosis. Through scaffold hopping, bioisosteric replacement design, and structure-activity relationship optimization, we developed a series of highly potent FGFRs inhibitors, and the indazole-containing candidate compound <strong>A16</strong> showed potent kinase activity comparable to that of <strong>AZD4547</strong>. In addition, <strong>A16</strong> effectively suppressed the activation of lung fibroblasts and hepatic stellate cells (HSCs) induced by TGF-β1, leading to a reduction in collagen deposition. Notably, <strong>A16</strong> exhibited potent anti-fibrotic effects through the inhibition of the FGFR pathway <em>in vitro</em>. Compound <strong>A16</strong> also showed reasonable pharmacokinetic properties (<em>F</em> = 21.84 %) and favorable cardiac safety (hERG IC<sub>50</sub> > 20 μM). Moreover, in models of pulmonary fibrosis, <strong>A16</strong> ameliorated (in the prevention model) and reversed (in the treatment model) bleomycin-induced lung fibrosis, as well as mitigated inflammatory immune response in the lung. Furthermore, in the CCl<sub>4</sub>-induced liver fibrosis model, when <strong>A16</strong> was administrated orally at a dose of 30 mg/kg/day for 3 weeks, it effectively improved liver function, restored damaged liver structures, and reduced collagen deposition. Taken together, these results suggest that <strong>A16</strong> could be a potential drug candidate for the treatment of organ fibrosis.</div></div>\",\"PeriodicalId\":314,\"journal\":{\"name\":\"European Journal of Medicinal Chemistry\",\"volume\":\"285 \",\"pages\":\"Article 117232\"},\"PeriodicalIF\":5.9000,\"publicationDate\":\"2025-03-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"European Journal of Medicinal Chemistry\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0223523424011140\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/2 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Medicinal Chemistry","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0223523424011140","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/2 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
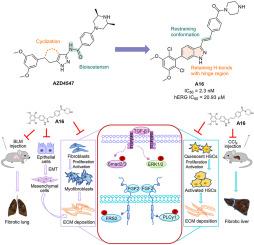
Design, synthesis, and biological evaluation of a potent and orally bioavailable FGFRs inhibitor for fibrotic treatment
Organ fibrosis, such as lung fibrosis and liver fibrosis, is a progressive and fatal disease. Fibroblast growth factor receptors (FGFRs) play an important role in the development and progression of fibrosis. Through scaffold hopping, bioisosteric replacement design, and structure-activity relationship optimization, we developed a series of highly potent FGFRs inhibitors, and the indazole-containing candidate compound A16 showed potent kinase activity comparable to that of AZD4547. In addition, A16 effectively suppressed the activation of lung fibroblasts and hepatic stellate cells (HSCs) induced by TGF-β1, leading to a reduction in collagen deposition. Notably, A16 exhibited potent anti-fibrotic effects through the inhibition of the FGFR pathway in vitro. Compound A16 also showed reasonable pharmacokinetic properties (F = 21.84 %) and favorable cardiac safety (hERG IC50 > 20 μM). Moreover, in models of pulmonary fibrosis, A16 ameliorated (in the prevention model) and reversed (in the treatment model) bleomycin-induced lung fibrosis, as well as mitigated inflammatory immune response in the lung. Furthermore, in the CCl4-induced liver fibrosis model, when A16 was administrated orally at a dose of 30 mg/kg/day for 3 weeks, it effectively improved liver function, restored damaged liver structures, and reduced collagen deposition. Taken together, these results suggest that A16 could be a potential drug candidate for the treatment of organ fibrosis.
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.




 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
