Thomas Hehre, Philip E. Klunzinger, Bernard Deppmeier, William Ohlinger, Warren Hehre
{"title":"实用机器学习策略。1 .修正MMFF分子力学模型,更准确地提供柔性有机分子的构象能差异","authors":"Thomas Hehre, Philip E. Klunzinger, Bernard Deppmeier, William Ohlinger, Warren Hehre","doi":"10.1002/jcc.70016","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>A correction to the MMFF molecular mechanics model, based on a neural network trained to reproduce conformer energy differences obtained from ωB97X-V/6-311+G(2df,2p)[6-311G*]//MMFF calculations is described. It is supported for molecules containing H, C, N, O, F, S, Cl, and Br. The correction adds only slightly to the cost of MMFF, and the resulting corrected model is several orders of magnitude faster than ωB97X-V/6-311+G(2df,2p)[6-311G*]. It properly identifies the lowest energy conformer for 82% of the molecules in a test set of flexible organic molecules (3553 total conformers), compared with 38% for MMFF. While the corrected MMFF model cannot be expected to provide sufficiently accurate Boltzmann weights for use in spectra and property calculations on flexible molecules, it is able to reduce the number of “reasonable” conformers that need to be passed on to more rigorous computational models, that can.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-01-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Practical Machine Learning Strategies. I. Correcting the MMFF Molecular Mechanics Model to More Accurately Provide Conformational Energy Differences in Flexible Organic Molecules\",\"authors\":\"Thomas Hehre, Philip E. Klunzinger, Bernard Deppmeier, William Ohlinger, Warren Hehre\",\"doi\":\"10.1002/jcc.70016\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n <p>A correction to the MMFF molecular mechanics model, based on a neural network trained to reproduce conformer energy differences obtained from ωB97X-V/6-311+G(2df,2p)[6-311G*]//MMFF calculations is described. It is supported for molecules containing H, C, N, O, F, S, Cl, and Br. The correction adds only slightly to the cost of MMFF, and the resulting corrected model is several orders of magnitude faster than ωB97X-V/6-311+G(2df,2p)[6-311G*]. It properly identifies the lowest energy conformer for 82% of the molecules in a test set of flexible organic molecules (3553 total conformers), compared with 38% for MMFF. While the corrected MMFF model cannot be expected to provide sufficiently accurate Boltzmann weights for use in spectra and property calculations on flexible molecules, it is able to reduce the number of “reasonable” conformers that need to be passed on to more rigorous computational models, that can.</p>\\n </div>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"46 1\",\"pages\":\"\"},\"PeriodicalIF\":4.8000,\"publicationDate\":\"2025-01-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70016\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70016","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Practical Machine Learning Strategies. I. Correcting the MMFF Molecular Mechanics Model to More Accurately Provide Conformational Energy Differences in Flexible Organic Molecules
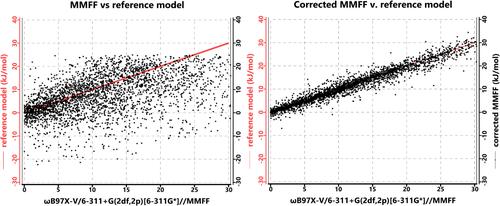
A correction to the MMFF molecular mechanics model, based on a neural network trained to reproduce conformer energy differences obtained from ωB97X-V/6-311+G(2df,2p)[6-311G*]//MMFF calculations is described. It is supported for molecules containing H, C, N, O, F, S, Cl, and Br. The correction adds only slightly to the cost of MMFF, and the resulting corrected model is several orders of magnitude faster than ωB97X-V/6-311+G(2df,2p)[6-311G*]. It properly identifies the lowest energy conformer for 82% of the molecules in a test set of flexible organic molecules (3553 total conformers), compared with 38% for MMFF. While the corrected MMFF model cannot be expected to provide sufficiently accurate Boltzmann weights for use in spectra and property calculations on flexible molecules, it is able to reduce the number of “reasonable” conformers that need to be passed on to more rigorous computational models, that can.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
