{"title":"用KNIME预测COX-2抑制剂:基于无代码自动机器学习的虚拟筛选工作流程","authors":"Powsali Ghosh, Ashok Kumar, Sushil Kumar Singh","doi":"10.1002/jcc.70030","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>Cyclooxygenase-2 (COX-2) is an enzyme that plays a crucial role in inflammation by converting arachidonic acid into prostaglandins. The overexpression of enzyme is associated with conditions such as cancer, arthritis, and Alzheimer's disease (AD), where it contributes to neuroinflammation. In silico virtual screening is pivotal in early-stage drug discovery; however, the absence of coding or machine learning expertise can impede the development of reliable computational models capable of accurately predicting inhibitor compounds based on their chemical structure. In this study, we developed an automated KNIME workflow for predicting the COX-2 inhibitory potential of novel molecules by building a multi-level ensemble model constructed with five machine learning algorithms (i.e., Logistic Regression, K-Nearest Neighbors, Decision Tree, Random Forest, and Extreme Gradient Boosting) and various molecular and fingerprint descriptors (i.e., AtomPair, Avalon, MACCS, Morgan, RDKit, and Pattern). Post-applicability domain filtering, the final majority voting-based ensemble model achieved 90.0% balanced accuracy, 87.7% precision, and 86.4% recall on the external validation set. The freely accessible workflow empowers users to swiftly and effortlessly predict COX-2 inhibitors, eliminating the need for any prior knowledge in machine learning, coding, or statistical modeling, significantly broadening its accessibility. While beginners can seamlessly use the tool as is, experienced KNIME users can leverage it as a foundation to build advanced workflows, driving further research and innovation.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 2","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-01-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"COX-2 Inhibitor Prediction With KNIME: A Codeless Automated Machine Learning-Based Virtual Screening Workflow\",\"authors\":\"Powsali Ghosh, Ashok Kumar, Sushil Kumar Singh\",\"doi\":\"10.1002/jcc.70030\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n <p>Cyclooxygenase-2 (COX-2) is an enzyme that plays a crucial role in inflammation by converting arachidonic acid into prostaglandins. The overexpression of enzyme is associated with conditions such as cancer, arthritis, and Alzheimer's disease (AD), where it contributes to neuroinflammation. In silico virtual screening is pivotal in early-stage drug discovery; however, the absence of coding or machine learning expertise can impede the development of reliable computational models capable of accurately predicting inhibitor compounds based on their chemical structure. In this study, we developed an automated KNIME workflow for predicting the COX-2 inhibitory potential of novel molecules by building a multi-level ensemble model constructed with five machine learning algorithms (i.e., Logistic Regression, K-Nearest Neighbors, Decision Tree, Random Forest, and Extreme Gradient Boosting) and various molecular and fingerprint descriptors (i.e., AtomPair, Avalon, MACCS, Morgan, RDKit, and Pattern). Post-applicability domain filtering, the final majority voting-based ensemble model achieved 90.0% balanced accuracy, 87.7% precision, and 86.4% recall on the external validation set. The freely accessible workflow empowers users to swiftly and effortlessly predict COX-2 inhibitors, eliminating the need for any prior knowledge in machine learning, coding, or statistical modeling, significantly broadening its accessibility. While beginners can seamlessly use the tool as is, experienced KNIME users can leverage it as a foundation to build advanced workflows, driving further research and innovation.</p>\\n </div>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"46 2\",\"pages\":\"\"},\"PeriodicalIF\":4.8000,\"publicationDate\":\"2025-01-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70030\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70030","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
COX-2 Inhibitor Prediction With KNIME: A Codeless Automated Machine Learning-Based Virtual Screening Workflow
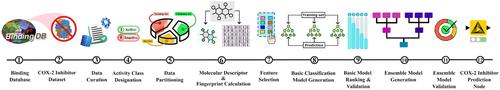
Cyclooxygenase-2 (COX-2) is an enzyme that plays a crucial role in inflammation by converting arachidonic acid into prostaglandins. The overexpression of enzyme is associated with conditions such as cancer, arthritis, and Alzheimer's disease (AD), where it contributes to neuroinflammation. In silico virtual screening is pivotal in early-stage drug discovery; however, the absence of coding or machine learning expertise can impede the development of reliable computational models capable of accurately predicting inhibitor compounds based on their chemical structure. In this study, we developed an automated KNIME workflow for predicting the COX-2 inhibitory potential of novel molecules by building a multi-level ensemble model constructed with five machine learning algorithms (i.e., Logistic Regression, K-Nearest Neighbors, Decision Tree, Random Forest, and Extreme Gradient Boosting) and various molecular and fingerprint descriptors (i.e., AtomPair, Avalon, MACCS, Morgan, RDKit, and Pattern). Post-applicability domain filtering, the final majority voting-based ensemble model achieved 90.0% balanced accuracy, 87.7% precision, and 86.4% recall on the external validation set. The freely accessible workflow empowers users to swiftly and effortlessly predict COX-2 inhibitors, eliminating the need for any prior knowledge in machine learning, coding, or statistical modeling, significantly broadening its accessibility. While beginners can seamlessly use the tool as is, experienced KNIME users can leverage it as a foundation to build advanced workflows, driving further research and innovation.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
