Jing-Ying Liu , Hong-En Zhang , Cheng Wang , Ping-Fan Zhang, Yun-Gen Xu, Lei Shi, Li-Ping Sun
{"title":"新型BRD4抑制剂三唑吡啶衍生物的设计、合成及抗肿瘤评价","authors":"Jing-Ying Liu , Hong-En Zhang , Cheng Wang , Ping-Fan Zhang, Yun-Gen Xu, Lei Shi, Li-Ping Sun","doi":"10.1016/j.ejmech.2025.117272","DOIUrl":null,"url":null,"abstract":"<div><div>The bromodomain-containing protein 4 (BRD4) is an epigenetic regulatory 'reader' belonging to the bromodomain and extra-terminal domain (BET) family. Several studies have demonstrated that the high expression of BRD4 is closely related to the occurrence and development of various cancers, so BRD4 has become a promising target for cancer treatment. However, there are no drugs targeting BRD4 available on the market, the development of novel BRD4 inhibitors is of great significance. This paper describes a series of triazolopyridine derivatives exhibiting favorable BRD4 inhibitory activity, which have not been reported before. Among them, the representative compound <strong>12m</strong> showed potent BRD4 BD1 inhibitory activity, of which the inhibition rate is better than the other compounds. In MV4-11 cell line, compound <strong>12m</strong> also showed excellent anti-cancer activity (IC<sub>50</sub> = 0.02 μM), which is superior to (+)-<strong>JQ1</strong> (IC<sub>50</sub> = 0.03 μM). Through molecular docking, it was discovered that compound <strong>12m</strong> could combine with the acetyl-lysine binding site of BRD4 BD1 and form a hydrogen bond with a crucial amino acid residue Asn140. Additionally, compound <strong>12m</strong> was found to have good metabolic stability with a clearance rate of only 0.3 μL/min/nm in mouse liver microsomes. Apoptosis experiments demonstrated that compound <strong>12m</strong> induced apoptosis better than (+)-<strong>JQ1</strong> at the same concentration, and the apoptosis rate of compound <strong>12m</strong> increased from 43.2 % to 83.2 %. Subsequent <em>in vivo</em> pharmacokinetic testing of compound <strong>12m</strong> in ICR mice yielded a good oral absorption and utilization of compound <strong>12m</strong> (F = 44.8 %). The results indicate that triazolopyridine is an outstanding skeleton for developing novel BRD4 inhibitors, and compound <strong>12m</strong> is a promising lead compound for further optimization and extensive clinical development.</div></div>","PeriodicalId":314,"journal":{"name":"European Journal of Medicinal Chemistry","volume":"285 ","pages":"Article 117272"},"PeriodicalIF":6.0000,"publicationDate":"2025-01-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Design, synthesis, and antitumor evaluation of triazolopyridine derivatives as novel inhibitors for BRD4\",\"authors\":\"Jing-Ying Liu , Hong-En Zhang , Cheng Wang , Ping-Fan Zhang, Yun-Gen Xu, Lei Shi, Li-Ping Sun\",\"doi\":\"10.1016/j.ejmech.2025.117272\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>The bromodomain-containing protein 4 (BRD4) is an epigenetic regulatory 'reader' belonging to the bromodomain and extra-terminal domain (BET) family. Several studies have demonstrated that the high expression of BRD4 is closely related to the occurrence and development of various cancers, so BRD4 has become a promising target for cancer treatment. However, there are no drugs targeting BRD4 available on the market, the development of novel BRD4 inhibitors is of great significance. This paper describes a series of triazolopyridine derivatives exhibiting favorable BRD4 inhibitory activity, which have not been reported before. Among them, the representative compound <strong>12m</strong> showed potent BRD4 BD1 inhibitory activity, of which the inhibition rate is better than the other compounds. In MV4-11 cell line, compound <strong>12m</strong> also showed excellent anti-cancer activity (IC<sub>50</sub> = 0.02 μM), which is superior to (+)-<strong>JQ1</strong> (IC<sub>50</sub> = 0.03 μM). Through molecular docking, it was discovered that compound <strong>12m</strong> could combine with the acetyl-lysine binding site of BRD4 BD1 and form a hydrogen bond with a crucial amino acid residue Asn140. Additionally, compound <strong>12m</strong> was found to have good metabolic stability with a clearance rate of only 0.3 μL/min/nm in mouse liver microsomes. Apoptosis experiments demonstrated that compound <strong>12m</strong> induced apoptosis better than (+)-<strong>JQ1</strong> at the same concentration, and the apoptosis rate of compound <strong>12m</strong> increased from 43.2 % to 83.2 %. Subsequent <em>in vivo</em> pharmacokinetic testing of compound <strong>12m</strong> in ICR mice yielded a good oral absorption and utilization of compound <strong>12m</strong> (F = 44.8 %). The results indicate that triazolopyridine is an outstanding skeleton for developing novel BRD4 inhibitors, and compound <strong>12m</strong> is a promising lead compound for further optimization and extensive clinical development.</div></div>\",\"PeriodicalId\":314,\"journal\":{\"name\":\"European Journal of Medicinal Chemistry\",\"volume\":\"285 \",\"pages\":\"Article 117272\"},\"PeriodicalIF\":6.0000,\"publicationDate\":\"2025-01-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"European Journal of Medicinal Chemistry\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0223523425000376\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Medicinal Chemistry","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0223523425000376","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Design, synthesis, and antitumor evaluation of triazolopyridine derivatives as novel inhibitors for BRD4
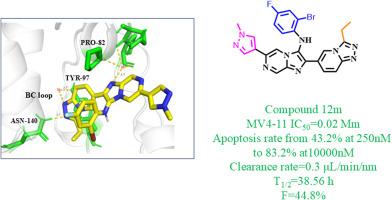
The bromodomain-containing protein 4 (BRD4) is an epigenetic regulatory 'reader' belonging to the bromodomain and extra-terminal domain (BET) family. Several studies have demonstrated that the high expression of BRD4 is closely related to the occurrence and development of various cancers, so BRD4 has become a promising target for cancer treatment. However, there are no drugs targeting BRD4 available on the market, the development of novel BRD4 inhibitors is of great significance. This paper describes a series of triazolopyridine derivatives exhibiting favorable BRD4 inhibitory activity, which have not been reported before. Among them, the representative compound 12m showed potent BRD4 BD1 inhibitory activity, of which the inhibition rate is better than the other compounds. In MV4-11 cell line, compound 12m also showed excellent anti-cancer activity (IC50 = 0.02 μM), which is superior to (+)-JQ1 (IC50 = 0.03 μM). Through molecular docking, it was discovered that compound 12m could combine with the acetyl-lysine binding site of BRD4 BD1 and form a hydrogen bond with a crucial amino acid residue Asn140. Additionally, compound 12m was found to have good metabolic stability with a clearance rate of only 0.3 μL/min/nm in mouse liver microsomes. Apoptosis experiments demonstrated that compound 12m induced apoptosis better than (+)-JQ1 at the same concentration, and the apoptosis rate of compound 12m increased from 43.2 % to 83.2 %. Subsequent in vivo pharmacokinetic testing of compound 12m in ICR mice yielded a good oral absorption and utilization of compound 12m (F = 44.8 %). The results indicate that triazolopyridine is an outstanding skeleton for developing novel BRD4 inhibitors, and compound 12m is a promising lead compound for further optimization and extensive clinical development.
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
