Ayush Shivhare, Bharti Dehariya, Shridhar R. Gadre and Milind M. Deshmukh
{"title":"循环协同性的贡献决定了分子簇中的氢键强度","authors":"Ayush Shivhare, Bharti Dehariya, Shridhar R. Gadre and Milind M. Deshmukh","doi":"10.1039/D4CP04741A","DOIUrl":null,"url":null,"abstract":"<p >In a recent communication (A. Shivhare, B. Dehariya, S. R. Gadre and M. M. Deshmukh, <em>Phys. Chem. Chem. Phys.</em>, 2024, <strong>26</strong>, 21332), we proposed a method for calculating the energy of a hydrogen bond (HB), which is common to two or more cyclic networks of HBs in water (H<small><sub>2</sub></small>O)<small><sub><em>n</em></sub></small> clusters. For this purpose, the sum of the cooperativity contributions (CCs) of these cyclic structures, estimated using the molecular tailoring approach (MTA)-based method, was added to the energy of this HB in the respective water dimer isolated from the cluster. The HB energies calculated in this fashion (<em>E</em><small><sup>Synergetic</sup></small><small><sub>HB</sub></small>) were in excellent agreement with their actual cluster counterparts (<em>E</em><small><sup>cluster</sup></small><small><sub>HB</sub></small>). In this work, we test the generality of this methodology. For this purpose, we employed clusters of ammonia <strong>(NH<small><sub>3</sub></small>)<small><sub><em>n</em></sub></small></strong>, hydrogen sulphide <strong>(H<small><sub>2</sub></small>S)<small><sub><em>n</em></sub></small></strong>, mixed <strong>(H<small><sub>2</sub></small>S)<small><sub><em>m</em></sub></small>(H<small><sub>2</sub></small>O)<small><sub><em>n</em></sub></small></strong>, <strong>(NH<small><sub>3</sub></small>)<small><sub><em>m</em></sub></small>(H<small><sub>2</sub></small>O)<small><sub><em>n</em></sub></small></strong>, methanol–water <strong>(CH<small><sub>3</sub></small>OH)<small><sub><em>m</em></sub></small>(H<small><sub>2</sub></small>O)<small><sub><em>n</em></sub></small></strong>, and hydrogen fluoride–water <strong>(HF)<small><sub><em>m</em></sub></small>(H<small><sub>2</sub></small>O)<small><sub><em>n</em></sub></small></strong> exhibiting HBs of variable strength (1 to 19 kcal mol<small><sup>−1</sup></small>). The HB energies in all these molecular clusters calculated using the present method were found to be accurate. The absolute difference between the <em>E</em><small><sup>Synergetic</sup></small><small><sub>HB</sub></small> and <em>E</em><small><sup>cluster</sup></small><small><sub>HB</sub></small> values in these clusters is found to be less than 0.5 kcal mol<small><sup>−1</sup></small>. Importantly, the present method not only enables accurate HB energy estimation in molecular clusters, but also offers qualitative guidelines for this purpose. The latter are based on the nature of cyclic cooperativity, exhibiting either full cyclic cooperativity (FCC), partial cyclic cooperativity (PCC) or anti-cooperativity (AC).</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 7","pages":" 3661-3672"},"PeriodicalIF":2.9000,"publicationDate":"2025-01-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Cyclic cooperativity contributions determine the hydrogen bond strengths in molecular clusters†\",\"authors\":\"Ayush Shivhare, Bharti Dehariya, Shridhar R. Gadre and Milind M. Deshmukh\",\"doi\":\"10.1039/D4CP04741A\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >In a recent communication (A. Shivhare, B. Dehariya, S. R. Gadre and M. M. Deshmukh, <em>Phys. Chem. Chem. Phys.</em>, 2024, <strong>26</strong>, 21332), we proposed a method for calculating the energy of a hydrogen bond (HB), which is common to two or more cyclic networks of HBs in water (H<small><sub>2</sub></small>O)<small><sub><em>n</em></sub></small> clusters. For this purpose, the sum of the cooperativity contributions (CCs) of these cyclic structures, estimated using the molecular tailoring approach (MTA)-based method, was added to the energy of this HB in the respective water dimer isolated from the cluster. The HB energies calculated in this fashion (<em>E</em><small><sup>Synergetic</sup></small><small><sub>HB</sub></small>) were in excellent agreement with their actual cluster counterparts (<em>E</em><small><sup>cluster</sup></small><small><sub>HB</sub></small>). In this work, we test the generality of this methodology. For this purpose, we employed clusters of ammonia <strong>(NH<small><sub>3</sub></small>)<small><sub><em>n</em></sub></small></strong>, hydrogen sulphide <strong>(H<small><sub>2</sub></small>S)<small><sub><em>n</em></sub></small></strong>, mixed <strong>(H<small><sub>2</sub></small>S)<small><sub><em>m</em></sub></small>(H<small><sub>2</sub></small>O)<small><sub><em>n</em></sub></small></strong>, <strong>(NH<small><sub>3</sub></small>)<small><sub><em>m</em></sub></small>(H<small><sub>2</sub></small>O)<small><sub><em>n</em></sub></small></strong>, methanol–water <strong>(CH<small><sub>3</sub></small>OH)<small><sub><em>m</em></sub></small>(H<small><sub>2</sub></small>O)<small><sub><em>n</em></sub></small></strong>, and hydrogen fluoride–water <strong>(HF)<small><sub><em>m</em></sub></small>(H<small><sub>2</sub></small>O)<small><sub><em>n</em></sub></small></strong> exhibiting HBs of variable strength (1 to 19 kcal mol<small><sup>−1</sup></small>). The HB energies in all these molecular clusters calculated using the present method were found to be accurate. The absolute difference between the <em>E</em><small><sup>Synergetic</sup></small><small><sub>HB</sub></small> and <em>E</em><small><sup>cluster</sup></small><small><sub>HB</sub></small> values in these clusters is found to be less than 0.5 kcal mol<small><sup>−1</sup></small>. Importantly, the present method not only enables accurate HB energy estimation in molecular clusters, but also offers qualitative guidelines for this purpose. The latter are based on the nature of cyclic cooperativity, exhibiting either full cyclic cooperativity (FCC), partial cyclic cooperativity (PCC) or anti-cooperativity (AC).</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 7\",\"pages\":\" 3661-3672\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-01-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp04741a\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp04741a","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Cyclic cooperativity contributions determine the hydrogen bond strengths in molecular clusters†
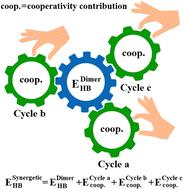
In a recent communication (A. Shivhare, B. Dehariya, S. R. Gadre and M. M. Deshmukh, Phys. Chem. Chem. Phys., 2024, 26, 21332), we proposed a method for calculating the energy of a hydrogen bond (HB), which is common to two or more cyclic networks of HBs in water (H2O)n clusters. For this purpose, the sum of the cooperativity contributions (CCs) of these cyclic structures, estimated using the molecular tailoring approach (MTA)-based method, was added to the energy of this HB in the respective water dimer isolated from the cluster. The HB energies calculated in this fashion (ESynergeticHB) were in excellent agreement with their actual cluster counterparts (EclusterHB). In this work, we test the generality of this methodology. For this purpose, we employed clusters of ammonia (NH3)n, hydrogen sulphide (H2S)n, mixed (H2S)m(H2O)n, (NH3)m(H2O)n, methanol–water (CH3OH)m(H2O)n, and hydrogen fluoride–water (HF)m(H2O)n exhibiting HBs of variable strength (1 to 19 kcal mol−1). The HB energies in all these molecular clusters calculated using the present method were found to be accurate. The absolute difference between the ESynergeticHB and EclusterHB values in these clusters is found to be less than 0.5 kcal mol−1. Importantly, the present method not only enables accurate HB energy estimation in molecular clusters, but also offers qualitative guidelines for this purpose. The latter are based on the nature of cyclic cooperativity, exhibiting either full cyclic cooperativity (FCC), partial cyclic cooperativity (PCC) or anti-cooperativity (AC).
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
