Róisín M. McManus, Max P. Komes, Angelika Griep, Francesco Santarelli, Stephanie Schwartz, Juan Ramón Perea, Johannes C.M. Schlachetzki, David S. Bouvier, Michelle-Amirah Khalil, Mario A. Lauterbach, Lea Heinemann, Titus Schlüter, Mehran Shaban Pour, Marta Lovotti, Rainer Stahl, Fraser Duthie, Juan F. Rodríguez-Alcázar, Susanne V. Schmidt, Jasper Spitzer, Peri Noori, Michael T. Heneka
{"title":"nlrp3介导的谷氨酰胺溶解控制小胶质细胞吞噬促进阿尔茨海默病进展","authors":"Róisín M. McManus, Max P. Komes, Angelika Griep, Francesco Santarelli, Stephanie Schwartz, Juan Ramón Perea, Johannes C.M. Schlachetzki, David S. Bouvier, Michelle-Amirah Khalil, Mario A. Lauterbach, Lea Heinemann, Titus Schlüter, Mehran Shaban Pour, Marta Lovotti, Rainer Stahl, Fraser Duthie, Juan F. Rodríguez-Alcázar, Susanne V. Schmidt, Jasper Spitzer, Peri Noori, Michael T. Heneka","doi":"10.1016/j.immuni.2025.01.007","DOIUrl":null,"url":null,"abstract":"Activation of the NLRP3 inflammasome has been implicated in the pathogenesis of Alzheimer’s disease (AD) via the release of IL-1β and ASC specks. However, whether NLRP3 is involved in pathways beyond this remained unknown. Here, we found that Aβ deposition <em>in vivo</em> directly triggered NLRP3 activation in APP/PS1 mice, which model many features of AD. Loss of NLRP3 increased glutamine- and glutamate-related metabolism and increased expression of microglial <em>Slc1a3</em>, which was associated with enhanced mitochondrial and metabolic activity. The generation of α-ketoglutarate during this process impacted cellular function, including increased clearance of Aβ peptides as well as epigenetic and gene transcription changes. This pathway was conserved between murine and human cells. Critically, we could mimic this effect pharmacologically using NLRP3-specific inhibitors, but only with chronic NLRP3 inhibition. Together, these data demonstrate an additional role for NLRP3, where it can modulate mitochondrial and metabolic function, with important downstream consequences for the progression of AD.","PeriodicalId":13269,"journal":{"name":"Immunity","volume":"52 1","pages":""},"PeriodicalIF":26.3000,"publicationDate":"2025-02-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"NLRP3-mediated glutaminolysis controls microglial phagocytosis to promote Alzheimer’s disease progression\",\"authors\":\"Róisín M. McManus, Max P. Komes, Angelika Griep, Francesco Santarelli, Stephanie Schwartz, Juan Ramón Perea, Johannes C.M. Schlachetzki, David S. Bouvier, Michelle-Amirah Khalil, Mario A. Lauterbach, Lea Heinemann, Titus Schlüter, Mehran Shaban Pour, Marta Lovotti, Rainer Stahl, Fraser Duthie, Juan F. Rodríguez-Alcázar, Susanne V. Schmidt, Jasper Spitzer, Peri Noori, Michael T. Heneka\",\"doi\":\"10.1016/j.immuni.2025.01.007\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Activation of the NLRP3 inflammasome has been implicated in the pathogenesis of Alzheimer’s disease (AD) via the release of IL-1β and ASC specks. However, whether NLRP3 is involved in pathways beyond this remained unknown. Here, we found that Aβ deposition <em>in vivo</em> directly triggered NLRP3 activation in APP/PS1 mice, which model many features of AD. Loss of NLRP3 increased glutamine- and glutamate-related metabolism and increased expression of microglial <em>Slc1a3</em>, which was associated with enhanced mitochondrial and metabolic activity. The generation of α-ketoglutarate during this process impacted cellular function, including increased clearance of Aβ peptides as well as epigenetic and gene transcription changes. This pathway was conserved between murine and human cells. Critically, we could mimic this effect pharmacologically using NLRP3-specific inhibitors, but only with chronic NLRP3 inhibition. Together, these data demonstrate an additional role for NLRP3, where it can modulate mitochondrial and metabolic function, with important downstream consequences for the progression of AD.\",\"PeriodicalId\":13269,\"journal\":{\"name\":\"Immunity\",\"volume\":\"52 1\",\"pages\":\"\"},\"PeriodicalIF\":26.3000,\"publicationDate\":\"2025-02-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Immunity\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1016/j.immuni.2025.01.007\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Immunity","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1016/j.immuni.2025.01.007","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
NLRP3-mediated glutaminolysis controls microglial phagocytosis to promote Alzheimer’s disease progression
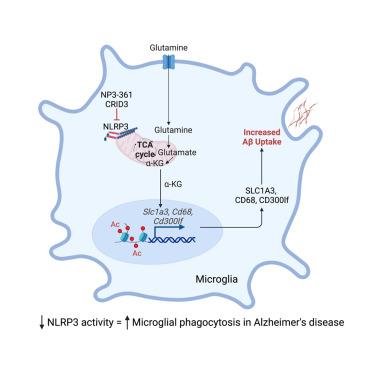
Activation of the NLRP3 inflammasome has been implicated in the pathogenesis of Alzheimer’s disease (AD) via the release of IL-1β and ASC specks. However, whether NLRP3 is involved in pathways beyond this remained unknown. Here, we found that Aβ deposition in vivo directly triggered NLRP3 activation in APP/PS1 mice, which model many features of AD. Loss of NLRP3 increased glutamine- and glutamate-related metabolism and increased expression of microglial Slc1a3, which was associated with enhanced mitochondrial and metabolic activity. The generation of α-ketoglutarate during this process impacted cellular function, including increased clearance of Aβ peptides as well as epigenetic and gene transcription changes. This pathway was conserved between murine and human cells. Critically, we could mimic this effect pharmacologically using NLRP3-specific inhibitors, but only with chronic NLRP3 inhibition. Together, these data demonstrate an additional role for NLRP3, where it can modulate mitochondrial and metabolic function, with important downstream consequences for the progression of AD.
期刊介绍:
Immunity is a publication that focuses on publishing significant advancements in research related to immunology. We encourage the submission of studies that offer groundbreaking immunological discoveries, whether at the molecular, cellular, or whole organism level. Topics of interest encompass a wide range, such as cancer, infectious diseases, neuroimmunology, autoimmune diseases, allergies, mucosal immunity, metabolic diseases, and homeostasis.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
