Peter Poliak, Patrick Bleiziffer, Felix Pultar, Sereina Riniker, Chris Oostenbrink
{"title":"用于GROMOS分子动力学的健壮和通用QM/MM接口","authors":"Peter Poliak, Patrick Bleiziffer, Felix Pultar, Sereina Riniker, Chris Oostenbrink","doi":"10.1002/jcc.70053","DOIUrl":null,"url":null,"abstract":"<p>The integration of quantum mechanics and molecular mechanics (QM/MM) within molecular dynamics simulations is crucial to accurately model complex biochemical systems. Here, we present an enhanced implementation of the QM/MM interface in the GROMOS simulation package, introducing significant improvements in functionality and user control. We present new features, including the link atom scheme, which allows the modeling of QM regions as a part of bigger molecules. Benchmark tests on various systems, including QM water in water, amino acids in water, and tripeptides validate the reliability of the new functionalities. Performance evaluations demonstrate that the updated implementation is efficient, with the primary computational burden attributed to the QM program rather than the QM/MM interface or the MD program itself. The improved QM/MM interface enables more advanced investigations into biomolecular reactivity, enzyme catalysis, and other phenomena requiring detailed quantum mechanical treatment within classical simulations. This work represents a significant advancement in the capabilities of GROMOS, providing enhanced tools to explore complex molecular systems.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 5","pages":""},"PeriodicalIF":2.9000,"publicationDate":"2025-02-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.70053","citationCount":"0","resultStr":"{\"title\":\"A Robust and Versatile QM/MM Interface for Molecular Dynamics in GROMOS\",\"authors\":\"Peter Poliak, Patrick Bleiziffer, Felix Pultar, Sereina Riniker, Chris Oostenbrink\",\"doi\":\"10.1002/jcc.70053\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The integration of quantum mechanics and molecular mechanics (QM/MM) within molecular dynamics simulations is crucial to accurately model complex biochemical systems. Here, we present an enhanced implementation of the QM/MM interface in the GROMOS simulation package, introducing significant improvements in functionality and user control. We present new features, including the link atom scheme, which allows the modeling of QM regions as a part of bigger molecules. Benchmark tests on various systems, including QM water in water, amino acids in water, and tripeptides validate the reliability of the new functionalities. Performance evaluations demonstrate that the updated implementation is efficient, with the primary computational burden attributed to the QM program rather than the QM/MM interface or the MD program itself. The improved QM/MM interface enables more advanced investigations into biomolecular reactivity, enzyme catalysis, and other phenomena requiring detailed quantum mechanical treatment within classical simulations. This work represents a significant advancement in the capabilities of GROMOS, providing enhanced tools to explore complex molecular systems.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"46 5\",\"pages\":\"\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-02-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.70053\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70053\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70053","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
A Robust and Versatile QM/MM Interface for Molecular Dynamics in GROMOS
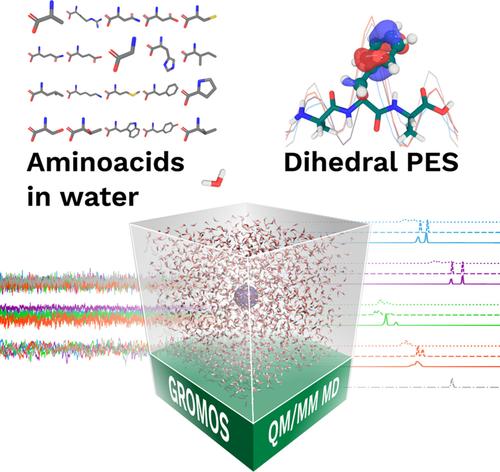
The integration of quantum mechanics and molecular mechanics (QM/MM) within molecular dynamics simulations is crucial to accurately model complex biochemical systems. Here, we present an enhanced implementation of the QM/MM interface in the GROMOS simulation package, introducing significant improvements in functionality and user control. We present new features, including the link atom scheme, which allows the modeling of QM regions as a part of bigger molecules. Benchmark tests on various systems, including QM water in water, amino acids in water, and tripeptides validate the reliability of the new functionalities. Performance evaluations demonstrate that the updated implementation is efficient, with the primary computational burden attributed to the QM program rather than the QM/MM interface or the MD program itself. The improved QM/MM interface enables more advanced investigations into biomolecular reactivity, enzyme catalysis, and other phenomena requiring detailed quantum mechanical treatment within classical simulations. This work represents a significant advancement in the capabilities of GROMOS, providing enhanced tools to explore complex molecular systems.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.




 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
