Rebaz Obaid Kareem, Yousif Hussein Azeez, Rebaz A. Omer, Karzan Mahmood Ahmed, Wali M. Hamad, Shalaw K. Salih, Dyari Mustafa Mamad
{"title":"合成2,4-二(4′-n-pentyloxybenzoyloxy)-苄基苯胺-4″-烷氧基苯胺的潜在缓蚀性能的联合DFT和Monte Carlo模拟研究","authors":"Rebaz Obaid Kareem, Yousif Hussein Azeez, Rebaz A. Omer, Karzan Mahmood Ahmed, Wali M. Hamad, Shalaw K. Salih, Dyari Mustafa Mamad","doi":"10.1002/slct.202403838","DOIUrl":null,"url":null,"abstract":"<p>This study explores the corrosion inhibition performance of novel organic compounds (C1–C10) on the Fe (110) metal surface using density functional theory (DFT) and Monte Carlo simulations (MC). The electronic properties, including HOMO (−5.209 to −5.294 eV), LUMO (−1.458 to −1.483 eV), and energy gaps (3.734–3.811 eV), were calculated. Compounds with lower energy gaps, such as C4 (3.734 eV), exhibited higher reactivity due to enhanced electron-donating ability. Global hardness (1.867–1.906 eV) and softness (0.262–0.268 eV⁻¹) further confirmed stability trends. Electron transfer (<i>ΔN</i>: 1.770–1.790) and back-donation energies from (−0.467 to −0.476 eV) indicated strong inhibitor-metal interactions. Monte Carlo simulations revealed adsorption energies ranging from −143.821 to −184.859 kcal/mol, confirming spontaneous adsorption. C10 demonstrated the highest adsorption energy (−184.859 kcal/mol) indicating superior stability and inhibition efficiency. The systematic addition of ─CH3 groups influenced electronic properties and adsorption behavior, with longer alkyl chains enhancing hydrophobic protection and electron-donating effects. The dipole moment values of the studied compounds (C1–C10) increase significantly from the gas phase to the aqueous phase (DMSO) indicating enhanced polarity and improved solubility in polar solvents. This study provides insights into designing effective corrosion inhibitors and highlights the utility of computational methods in predicting their performance.</p>","PeriodicalId":146,"journal":{"name":"ChemistrySelect","volume":"10 13","pages":""},"PeriodicalIF":2.0000,"publicationDate":"2025-03-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Combined DFT and Monte Carlo Simulation Studies of Potential Corrosion Inhibition Properties of Synthesis 2,4-bis(4′-n-pentyloxybenzoyloxy)-benzylidine-4″-nalkoxyaniline\",\"authors\":\"Rebaz Obaid Kareem, Yousif Hussein Azeez, Rebaz A. Omer, Karzan Mahmood Ahmed, Wali M. Hamad, Shalaw K. Salih, Dyari Mustafa Mamad\",\"doi\":\"10.1002/slct.202403838\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>This study explores the corrosion inhibition performance of novel organic compounds (C1–C10) on the Fe (110) metal surface using density functional theory (DFT) and Monte Carlo simulations (MC). The electronic properties, including HOMO (−5.209 to −5.294 eV), LUMO (−1.458 to −1.483 eV), and energy gaps (3.734–3.811 eV), were calculated. Compounds with lower energy gaps, such as C4 (3.734 eV), exhibited higher reactivity due to enhanced electron-donating ability. Global hardness (1.867–1.906 eV) and softness (0.262–0.268 eV⁻¹) further confirmed stability trends. Electron transfer (<i>ΔN</i>: 1.770–1.790) and back-donation energies from (−0.467 to −0.476 eV) indicated strong inhibitor-metal interactions. Monte Carlo simulations revealed adsorption energies ranging from −143.821 to −184.859 kcal/mol, confirming spontaneous adsorption. C10 demonstrated the highest adsorption energy (−184.859 kcal/mol) indicating superior stability and inhibition efficiency. The systematic addition of ─CH3 groups influenced electronic properties and adsorption behavior, with longer alkyl chains enhancing hydrophobic protection and electron-donating effects. The dipole moment values of the studied compounds (C1–C10) increase significantly from the gas phase to the aqueous phase (DMSO) indicating enhanced polarity and improved solubility in polar solvents. This study provides insights into designing effective corrosion inhibitors and highlights the utility of computational methods in predicting their performance.</p>\",\"PeriodicalId\":146,\"journal\":{\"name\":\"ChemistrySelect\",\"volume\":\"10 13\",\"pages\":\"\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2025-03-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ChemistrySelect\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/slct.202403838\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ChemistrySelect","FirstCategoryId":"92","ListUrlMain":"https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/slct.202403838","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Combined DFT and Monte Carlo Simulation Studies of Potential Corrosion Inhibition Properties of Synthesis 2,4-bis(4′-n-pentyloxybenzoyloxy)-benzylidine-4″-nalkoxyaniline
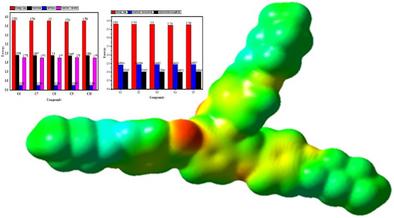
This study explores the corrosion inhibition performance of novel organic compounds (C1–C10) on the Fe (110) metal surface using density functional theory (DFT) and Monte Carlo simulations (MC). The electronic properties, including HOMO (−5.209 to −5.294 eV), LUMO (−1.458 to −1.483 eV), and energy gaps (3.734–3.811 eV), were calculated. Compounds with lower energy gaps, such as C4 (3.734 eV), exhibited higher reactivity due to enhanced electron-donating ability. Global hardness (1.867–1.906 eV) and softness (0.262–0.268 eV⁻¹) further confirmed stability trends. Electron transfer (ΔN: 1.770–1.790) and back-donation energies from (−0.467 to −0.476 eV) indicated strong inhibitor-metal interactions. Monte Carlo simulations revealed adsorption energies ranging from −143.821 to −184.859 kcal/mol, confirming spontaneous adsorption. C10 demonstrated the highest adsorption energy (−184.859 kcal/mol) indicating superior stability and inhibition efficiency. The systematic addition of ─CH3 groups influenced electronic properties and adsorption behavior, with longer alkyl chains enhancing hydrophobic protection and electron-donating effects. The dipole moment values of the studied compounds (C1–C10) increase significantly from the gas phase to the aqueous phase (DMSO) indicating enhanced polarity and improved solubility in polar solvents. This study provides insights into designing effective corrosion inhibitors and highlights the utility of computational methods in predicting their performance.
期刊介绍:
ChemistrySelect is the latest journal from ChemPubSoc Europe and Wiley-VCH. It offers researchers a quality society-owned journal in which to publish their work in all areas of chemistry. Manuscripts are evaluated by active researchers to ensure they add meaningfully to the scientific literature, and those accepted are processed quickly to ensure rapid online publication.







 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
