Shidrokh Ardestani, Desirae L Deskins, Pampee P Young
{"title":"膜tnf - α激活的程序性坏死是由神经酰胺诱导的活性氧介导的。","authors":"Shidrokh Ardestani, Desirae L Deskins, Pampee P Young","doi":"10.1186/1750-2187-8-12","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Programmed necrosis is a form of caspase-independent cell death whose molecular regulation is poorly understood. While tumor necrosis factor-alpha (TNF-α) has been identified as an activator of programmed necrosis, the specific context under which this can happen is unclear. Recently we reported that TNF-α can be expressed by human tumor cells as both a membrane tethered (mTNF-α) and a soluble (sTNF-α) form. Whereas low level, tumor-derived sTNF-α acts as a tumor promoter, tumor cell expression of mTNF-α significantly delays tumor growth in mice, in large part by induction of programmed necrosis of tumor associated myeloid cells. In this study we sought to determine the molecular mechanism involved in mTNF-α oxidative stress-induced cell death by evaluating the known pathways involved in TNF receptor-induced programmed necrosis.</p><p><strong>Methods: </strong>The source of Reactive Oxygen Species (ROS) in mTNF-α treated cells was determined by coculturing RAW 264.7 monocytic and L929 fibroblasts cells with fixed B16F10 control or mTNF-α expressing-melanoma cells in the presence of inhibitors of NADPH and mitochondria ROS. To identify the down-stream effector of TNF-a receptors (TNFR), level of phospho-RIP-1 and ceramide activity were evaluated.To determine whether mTNF-mediated cell death was dependent on a specific TNFR, cell death was measured in primary CD11b myeloid cells isolated from wild-type or TNFR-1, TNFR-2, TNFR-1 and TNFR-2 double knockout mice, cocultured with various TNF-α isoform.</p><p><strong>Results: </strong>Tumor derived-mTNF-α increased ROS-mediated cytotoxicity, independent of caspase-3 activity. Although TNFR on target cells were required for this effect, we observed that mTNF-induced cell death could be mediated through both TNFR-1 and the death domain-lacking TNFR-2. ROS generation and cytotoxicity were inhibited by a mitochondrial respiratory chain inhibitor but not by an inhibitor of NADPH oxidase. mTNF-α mediated cytotoxicity was independent of RIP-1, a serine/threonine kinase that serves as a main adaptor protein of sTNF-α induced programmed necrosis. Instead, mTNF-α-induced ROS and cell death was prohibited by the ceramide-activated protein kinase (CAPK) inhibitor.</p><p><strong>Conclusion: </strong>These findings demonstrate that the mTNF-α isoform is an effective inducer of programmed necrosis through a caspase independent, ceramide-related pathway. Interestingly, unlike sTNFα, mTNF-induced programmed necrosis is not dependent on the presence of TNFR1.</p>","PeriodicalId":35051,"journal":{"name":"Journal of Molecular Signaling","volume":"8 1","pages":"12"},"PeriodicalIF":0.0000,"publicationDate":"2013-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3895838/pdf/","citationCount":"53","resultStr":"{\"title\":\"Membrane TNF-alpha-activated programmed necrosis is mediated by Ceramide-induced reactive oxygen species.\",\"authors\":\"Shidrokh Ardestani, Desirae L Deskins, Pampee P Young\",\"doi\":\"10.1186/1750-2187-8-12\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Programmed necrosis is a form of caspase-independent cell death whose molecular regulation is poorly understood. While tumor necrosis factor-alpha (TNF-α) has been identified as an activator of programmed necrosis, the specific context under which this can happen is unclear. Recently we reported that TNF-α can be expressed by human tumor cells as both a membrane tethered (mTNF-α) and a soluble (sTNF-α) form. Whereas low level, tumor-derived sTNF-α acts as a tumor promoter, tumor cell expression of mTNF-α significantly delays tumor growth in mice, in large part by induction of programmed necrosis of tumor associated myeloid cells. In this study we sought to determine the molecular mechanism involved in mTNF-α oxidative stress-induced cell death by evaluating the known pathways involved in TNF receptor-induced programmed necrosis.</p><p><strong>Methods: </strong>The source of Reactive Oxygen Species (ROS) in mTNF-α treated cells was determined by coculturing RAW 264.7 monocytic and L929 fibroblasts cells with fixed B16F10 control or mTNF-α expressing-melanoma cells in the presence of inhibitors of NADPH and mitochondria ROS. To identify the down-stream effector of TNF-a receptors (TNFR), level of phospho-RIP-1 and ceramide activity were evaluated.To determine whether mTNF-mediated cell death was dependent on a specific TNFR, cell death was measured in primary CD11b myeloid cells isolated from wild-type or TNFR-1, TNFR-2, TNFR-1 and TNFR-2 double knockout mice, cocultured with various TNF-α isoform.</p><p><strong>Results: </strong>Tumor derived-mTNF-α increased ROS-mediated cytotoxicity, independent of caspase-3 activity. Although TNFR on target cells were required for this effect, we observed that mTNF-induced cell death could be mediated through both TNFR-1 and the death domain-lacking TNFR-2. ROS generation and cytotoxicity were inhibited by a mitochondrial respiratory chain inhibitor but not by an inhibitor of NADPH oxidase. mTNF-α mediated cytotoxicity was independent of RIP-1, a serine/threonine kinase that serves as a main adaptor protein of sTNF-α induced programmed necrosis. Instead, mTNF-α-induced ROS and cell death was prohibited by the ceramide-activated protein kinase (CAPK) inhibitor.</p><p><strong>Conclusion: </strong>These findings demonstrate that the mTNF-α isoform is an effective inducer of programmed necrosis through a caspase independent, ceramide-related pathway. Interestingly, unlike sTNFα, mTNF-induced programmed necrosis is not dependent on the presence of TNFR1.</p>\",\"PeriodicalId\":35051,\"journal\":{\"name\":\"Journal of Molecular Signaling\",\"volume\":\"8 1\",\"pages\":\"12\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2013-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3895838/pdf/\",\"citationCount\":\"53\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Signaling\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/1750-2187-8-12\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Signaling","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1750-2187-8-12","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Membrane TNF-alpha-activated programmed necrosis is mediated by Ceramide-induced reactive oxygen species.
Background: Programmed necrosis is a form of caspase-independent cell death whose molecular regulation is poorly understood. While tumor necrosis factor-alpha (TNF-α) has been identified as an activator of programmed necrosis, the specific context under which this can happen is unclear. Recently we reported that TNF-α can be expressed by human tumor cells as both a membrane tethered (mTNF-α) and a soluble (sTNF-α) form. Whereas low level, tumor-derived sTNF-α acts as a tumor promoter, tumor cell expression of mTNF-α significantly delays tumor growth in mice, in large part by induction of programmed necrosis of tumor associated myeloid cells. In this study we sought to determine the molecular mechanism involved in mTNF-α oxidative stress-induced cell death by evaluating the known pathways involved in TNF receptor-induced programmed necrosis.
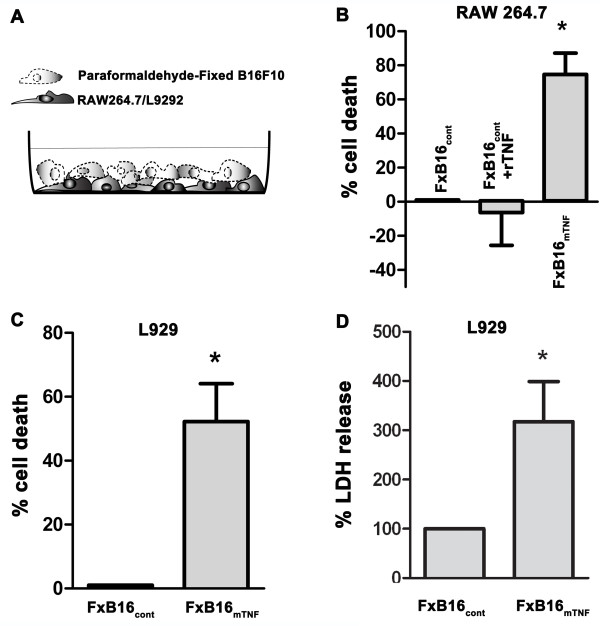
Methods: The source of Reactive Oxygen Species (ROS) in mTNF-α treated cells was determined by coculturing RAW 264.7 monocytic and L929 fibroblasts cells with fixed B16F10 control or mTNF-α expressing-melanoma cells in the presence of inhibitors of NADPH and mitochondria ROS. To identify the down-stream effector of TNF-a receptors (TNFR), level of phospho-RIP-1 and ceramide activity were evaluated.To determine whether mTNF-mediated cell death was dependent on a specific TNFR, cell death was measured in primary CD11b myeloid cells isolated from wild-type or TNFR-1, TNFR-2, TNFR-1 and TNFR-2 double knockout mice, cocultured with various TNF-α isoform.
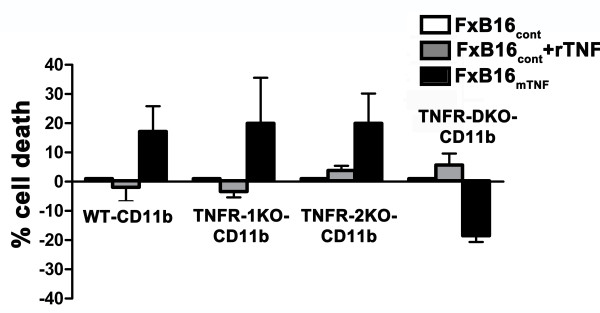
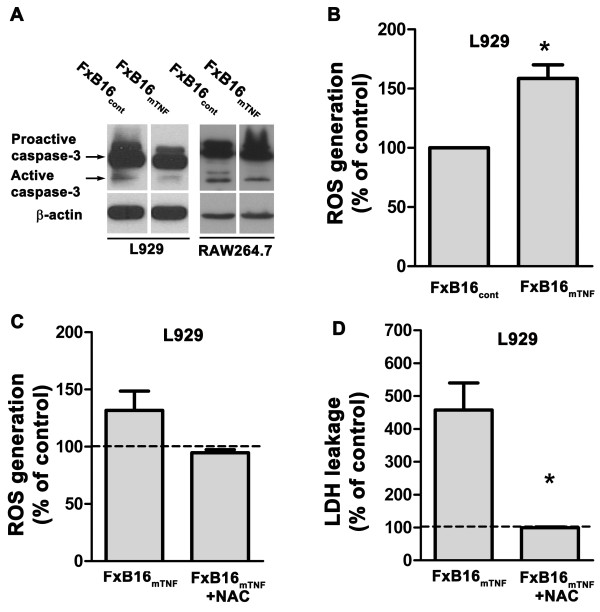
Results: Tumor derived-mTNF-α increased ROS-mediated cytotoxicity, independent of caspase-3 activity. Although TNFR on target cells were required for this effect, we observed that mTNF-induced cell death could be mediated through both TNFR-1 and the death domain-lacking TNFR-2. ROS generation and cytotoxicity were inhibited by a mitochondrial respiratory chain inhibitor but not by an inhibitor of NADPH oxidase. mTNF-α mediated cytotoxicity was independent of RIP-1, a serine/threonine kinase that serves as a main adaptor protein of sTNF-α induced programmed necrosis. Instead, mTNF-α-induced ROS and cell death was prohibited by the ceramide-activated protein kinase (CAPK) inhibitor.
Conclusion: These findings demonstrate that the mTNF-α isoform is an effective inducer of programmed necrosis through a caspase independent, ceramide-related pathway. Interestingly, unlike sTNFα, mTNF-induced programmed necrosis is not dependent on the presence of TNFR1.
期刊介绍:
Journal of Molecular Signaling is an open access, peer-reviewed online journal that encompasses all aspects of molecular signaling. Molecular signaling is an exponentially growing field that encompasses different molecular aspects of cell signaling underlying normal and pathological conditions. Specifically, the research area of the journal is on the normal or aberrant molecular mechanisms involving receptors, G-proteins, kinases, phosphatases, and transcription factors in regulating cell proliferation, differentiation, apoptosis, and oncogenesis in mammalian cells. This area also covers the genetic and epigenetic changes that modulate the signaling properties of cells and the resultant physiological conditions.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
