Doglas Parise, Mariana T D Parise, Marcus V C Viana, Adrian V Muñoz-Bucio, Yazmin A Cortés-Pérez, Beatriz Arellano-Reynoso, Efrén Díaz-Aparicio, Fernanda A Dorella, Felipe L Pereira, Alex F Carvalho, Henrique C P Figueiredo, Preetam Ghosh, Debmalya Barh, Anne C P Gomide, Vasco A C Azevedo
{"title":"墨西哥假结核棒状杆菌菌株的首次基因组测序和比较分析。","authors":"Doglas Parise, Mariana T D Parise, Marcus V C Viana, Adrian V Muñoz-Bucio, Yazmin A Cortés-Pérez, Beatriz Arellano-Reynoso, Efrén Díaz-Aparicio, Fernanda A Dorella, Felipe L Pereira, Alex F Carvalho, Henrique C P Figueiredo, Preetam Ghosh, Debmalya Barh, Anne C P Gomide, Vasco A C Azevedo","doi":"10.1186/s40793-018-0325-z","DOIUrl":null,"url":null,"abstract":"<p><p><i>Corynebacterium pseudotuberculosis</i> is a pathogenic bacterium which has been rapidly spreading all over the world, causing economic losses in the agricultural sector and sporadically infecting humans. Six <i>C. pseudotuberculosis</i> strains were isolated from goats, sheep, and horses with distinct abscess locations. For the first time, Mexican genomes of this bacterium were sequenced and studied in silico. All strains were sequenced using Ion Personal Genome Machine sequencer, assembled using Newbler and SPAdes software. The automatic genome annotation was done using the software RAST and in-house scripts for transference, followed by manual curation using Artemis software and BLAST against NCBI and UniProt databases. The six genomes are publicly available in NCBI database. The analysis of nucleotide sequence similarity and the generated phylogenetic tree led to the observation that the Mexican strains are more similar between strains from the same host, but the genetic structure is probably more influenced by transportation of animals between farms than host preference. Also, a putative drug target was predicted and in silico analysis of 46 strains showed two gene clusters capable of differentiating the biovars <i>equi</i> and <i>ovis</i>: Restriction Modification system and CRISPR-Cas cluster.</p>","PeriodicalId":21965,"journal":{"name":"Standards in Genomic Sciences","volume":"13 ","pages":"21"},"PeriodicalIF":0.0000,"publicationDate":"2018-10-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6180578/pdf/","citationCount":"0","resultStr":"{\"title\":\"First genome sequencing and comparative analyses of <i>Corynebacterium pseudotuberculosis</i> strains from Mexico.\",\"authors\":\"Doglas Parise, Mariana T D Parise, Marcus V C Viana, Adrian V Muñoz-Bucio, Yazmin A Cortés-Pérez, Beatriz Arellano-Reynoso, Efrén Díaz-Aparicio, Fernanda A Dorella, Felipe L Pereira, Alex F Carvalho, Henrique C P Figueiredo, Preetam Ghosh, Debmalya Barh, Anne C P Gomide, Vasco A C Azevedo\",\"doi\":\"10.1186/s40793-018-0325-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p><i>Corynebacterium pseudotuberculosis</i> is a pathogenic bacterium which has been rapidly spreading all over the world, causing economic losses in the agricultural sector and sporadically infecting humans. Six <i>C. pseudotuberculosis</i> strains were isolated from goats, sheep, and horses with distinct abscess locations. For the first time, Mexican genomes of this bacterium were sequenced and studied in silico. All strains were sequenced using Ion Personal Genome Machine sequencer, assembled using Newbler and SPAdes software. The automatic genome annotation was done using the software RAST and in-house scripts for transference, followed by manual curation using Artemis software and BLAST against NCBI and UniProt databases. The six genomes are publicly available in NCBI database. The analysis of nucleotide sequence similarity and the generated phylogenetic tree led to the observation that the Mexican strains are more similar between strains from the same host, but the genetic structure is probably more influenced by transportation of animals between farms than host preference. Also, a putative drug target was predicted and in silico analysis of 46 strains showed two gene clusters capable of differentiating the biovars <i>equi</i> and <i>ovis</i>: Restriction Modification system and CRISPR-Cas cluster.</p>\",\"PeriodicalId\":21965,\"journal\":{\"name\":\"Standards in Genomic Sciences\",\"volume\":\"13 \",\"pages\":\"21\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2018-10-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6180578/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Standards in Genomic Sciences\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s40793-018-0325-z\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2018/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Standards in Genomic Sciences","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s40793-018-0325-z","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 0
摘要
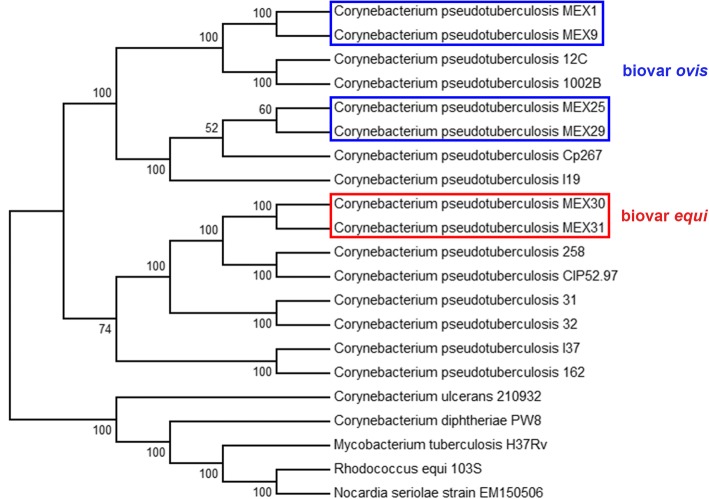
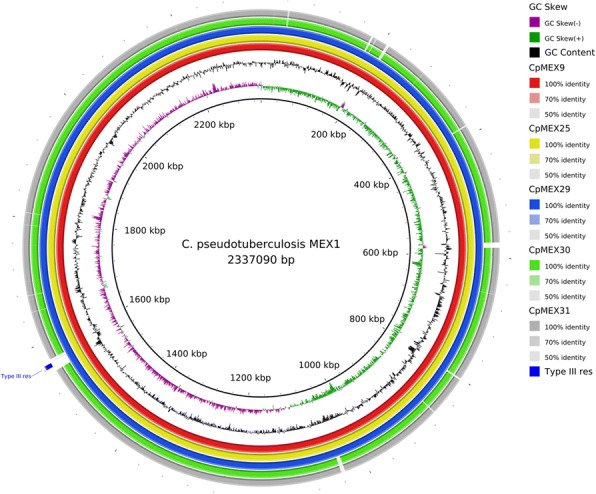
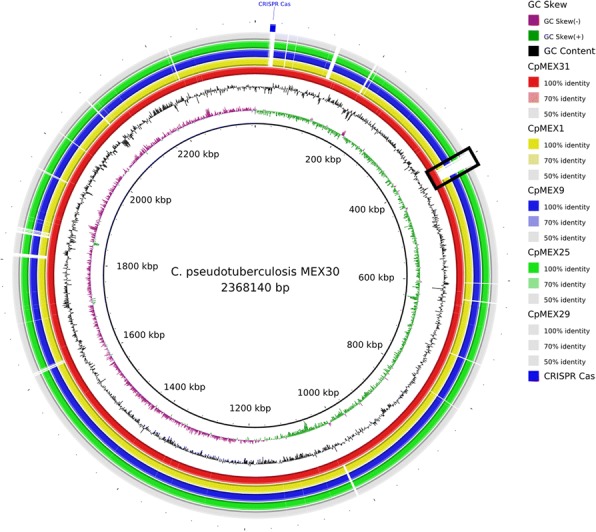
假结核棒状杆菌是一种病原菌,在全球迅速蔓延,给农业部门造成经济损失,并零星感染人类。从山羊、绵羊和马身上分离出了六株伪结核杆菌,它们的脓肿位置各不相同。首次对这种细菌的墨西哥基因组进行了测序和研究。所有菌株均使用 Ion Personal Genome Machine 测序仪进行测序,并使用 Newbler 和 SPAdes 软件进行组装。使用 RAST 软件和内部脚本进行了自动基因组注释,然后使用 Artemis 软件和 BLAST 对 NCBI 和 UniProt 数据库进行了人工整理。这六个基因组已在 NCBI 数据库中公开。对核苷酸序列相似性和生成的系统发生树进行分析后发现,墨西哥菌株与来自同一宿主的菌株之间更为相似,但遗传结构可能更多地受到农场之间动物运输的影响,而不是宿主偏好的影响。此外,还预测了一个可能的药物靶点,并对 46 株菌株进行了硅学分析,结果显示有两个基因簇能够区分马属生物和羱属生物:限制性修饰系统和CRISPR-Cas基因簇。
First genome sequencing and comparative analyses of Corynebacterium pseudotuberculosis strains from Mexico.
Corynebacterium pseudotuberculosis is a pathogenic bacterium which has been rapidly spreading all over the world, causing economic losses in the agricultural sector and sporadically infecting humans. Six C. pseudotuberculosis strains were isolated from goats, sheep, and horses with distinct abscess locations. For the first time, Mexican genomes of this bacterium were sequenced and studied in silico. All strains were sequenced using Ion Personal Genome Machine sequencer, assembled using Newbler and SPAdes software. The automatic genome annotation was done using the software RAST and in-house scripts for transference, followed by manual curation using Artemis software and BLAST against NCBI and UniProt databases. The six genomes are publicly available in NCBI database. The analysis of nucleotide sequence similarity and the generated phylogenetic tree led to the observation that the Mexican strains are more similar between strains from the same host, but the genetic structure is probably more influenced by transportation of animals between farms than host preference. Also, a putative drug target was predicted and in silico analysis of 46 strains showed two gene clusters capable of differentiating the biovars equi and ovis: Restriction Modification system and CRISPR-Cas cluster.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
