{"title":"增生Ulva对高温胁迫反应的比较蛋白质组学分析。","authors":"Meihua Fan, Xue Sun, Zhi Liao, Jianxin Wang, Yahe Li, Nianjun Xu","doi":"10.1186/s12953-018-0145-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong><i>Ulva prolifera</i> belongs to green macroalgae and is the dominant species of green tide. It is distributed worldwide and is therefore subject to high-temperature stress during the growth process. However, the adaptation mechanisms of the response of <i>U. prolifera</i> to high temperatures have not been clearly investigated yet.</p><p><strong>Methods: </strong>In this study, isobaric tags for relative and absolute quantitation (iTRAQ) labelling was applied in combination with the liquid chromatography-tandem mass spectrometry (LC-MS/MS) to conduct comparative proteomic analysis of the response of <i>U. prolifera</i> to high-temperature stress and to elucidate the involvement of this response in adaptation mechanisms. Differentially expressed proteins (DEPs) of <i>U. prolifera</i> under high temperature (denote UpHT) compared with the control (UpC) were identified. Bioinformatic analyses including GO analysis, pathway analysis, and pathway enrichment analysis was performed to analyse the key metabolic pathways that underlie the thermal tolerance mechanism through protein networks. Quantitative real-time PCR and western blot were performed to validate selected proteins.</p><p><strong>Results: </strong>In the present study, 1223 DEPs were identified under high temperature compared with the control, which included 790 up-regulated and 433 down-regulated proteins. The high-temperature stimulus mainly induced the expression of glutathione S-transferase, heat shock protein, ascorbate peroxidase, manganese superoxide dismutase, ubiquitin-related protein, lhcSR, rubisco activase, serine/threonine protein kinase 2, adenylate kinase, Ca<sup>2+</sup>-dependent protein kinase (CDPK), disease resistance protein EDS1, metacaspase type II, NDPK2a, 26S proteasome regulatory subunit, ubiquinone oxidoreductase, ATP synthase subunit, SnRK2s, and cytochrome P450. The down-regulated proteins were photosynthesis-related proteins, glutathione reductase, catalase-peroxidase, thioredoxin, thioredoxin peroxidase, PP2C, and carbon fixation-related proteins. Furthermore, biological index analysis indicated that protein content and SOD activity decreased; the value of Fv/Fm dropped to the lowest point after culture for 96 h. However, APX activity and MDA content increased under high temperature.</p><p><strong>Conclusion: </strong>The present study implied an increase in proteins that were associated with the stress response, oxidative phosphorylation, the cytokinin signal transduction pathway, the abscisic acid signal transduction pathway, and the glutathione metabolism pathway. Proteins that were associated with photosynthesis, carbon fixation in photosynthesis organisms, and the photosynthesis antenna protein pathway were decreased. These pathways played a pivotal role in high temperature regulation. These novel proteins provide a good starting point for further research into their functions using genetic or other approaches. These findings significantly improve the understanding of the molecular mechanisms involved in the tolerance of algae to high-temperature stress.</p>","PeriodicalId":20857,"journal":{"name":"Proteome Science","volume":"16 ","pages":"17"},"PeriodicalIF":1.6000,"publicationDate":"2018-10-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12953-018-0145-5","citationCount":"19","resultStr":"{\"title\":\"Comparative proteomic analysis of <i>Ulva prolifera</i> response to high temperature stress.\",\"authors\":\"Meihua Fan, Xue Sun, Zhi Liao, Jianxin Wang, Yahe Li, Nianjun Xu\",\"doi\":\"10.1186/s12953-018-0145-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong><i>Ulva prolifera</i> belongs to green macroalgae and is the dominant species of green tide. It is distributed worldwide and is therefore subject to high-temperature stress during the growth process. However, the adaptation mechanisms of the response of <i>U. prolifera</i> to high temperatures have not been clearly investigated yet.</p><p><strong>Methods: </strong>In this study, isobaric tags for relative and absolute quantitation (iTRAQ) labelling was applied in combination with the liquid chromatography-tandem mass spectrometry (LC-MS/MS) to conduct comparative proteomic analysis of the response of <i>U. prolifera</i> to high-temperature stress and to elucidate the involvement of this response in adaptation mechanisms. Differentially expressed proteins (DEPs) of <i>U. prolifera</i> under high temperature (denote UpHT) compared with the control (UpC) were identified. Bioinformatic analyses including GO analysis, pathway analysis, and pathway enrichment analysis was performed to analyse the key metabolic pathways that underlie the thermal tolerance mechanism through protein networks. Quantitative real-time PCR and western blot were performed to validate selected proteins.</p><p><strong>Results: </strong>In the present study, 1223 DEPs were identified under high temperature compared with the control, which included 790 up-regulated and 433 down-regulated proteins. The high-temperature stimulus mainly induced the expression of glutathione S-transferase, heat shock protein, ascorbate peroxidase, manganese superoxide dismutase, ubiquitin-related protein, lhcSR, rubisco activase, serine/threonine protein kinase 2, adenylate kinase, Ca<sup>2+</sup>-dependent protein kinase (CDPK), disease resistance protein EDS1, metacaspase type II, NDPK2a, 26S proteasome regulatory subunit, ubiquinone oxidoreductase, ATP synthase subunit, SnRK2s, and cytochrome P450. The down-regulated proteins were photosynthesis-related proteins, glutathione reductase, catalase-peroxidase, thioredoxin, thioredoxin peroxidase, PP2C, and carbon fixation-related proteins. Furthermore, biological index analysis indicated that protein content and SOD activity decreased; the value of Fv/Fm dropped to the lowest point after culture for 96 h. However, APX activity and MDA content increased under high temperature.</p><p><strong>Conclusion: </strong>The present study implied an increase in proteins that were associated with the stress response, oxidative phosphorylation, the cytokinin signal transduction pathway, the abscisic acid signal transduction pathway, and the glutathione metabolism pathway. Proteins that were associated with photosynthesis, carbon fixation in photosynthesis organisms, and the photosynthesis antenna protein pathway were decreased. These pathways played a pivotal role in high temperature regulation. These novel proteins provide a good starting point for further research into their functions using genetic or other approaches. These findings significantly improve the understanding of the molecular mechanisms involved in the tolerance of algae to high-temperature stress.</p>\",\"PeriodicalId\":20857,\"journal\":{\"name\":\"Proteome Science\",\"volume\":\"16 \",\"pages\":\"17\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2018-10-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s12953-018-0145-5\",\"citationCount\":\"19\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Proteome Science\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12953-018-0145-5\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2018/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteome Science","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12953-018-0145-5","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Comparative proteomic analysis of Ulva prolifera response to high temperature stress.
Background: Ulva prolifera belongs to green macroalgae and is the dominant species of green tide. It is distributed worldwide and is therefore subject to high-temperature stress during the growth process. However, the adaptation mechanisms of the response of U. prolifera to high temperatures have not been clearly investigated yet.
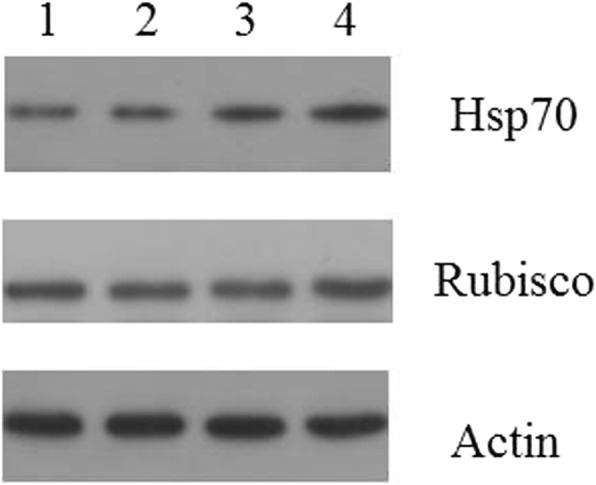
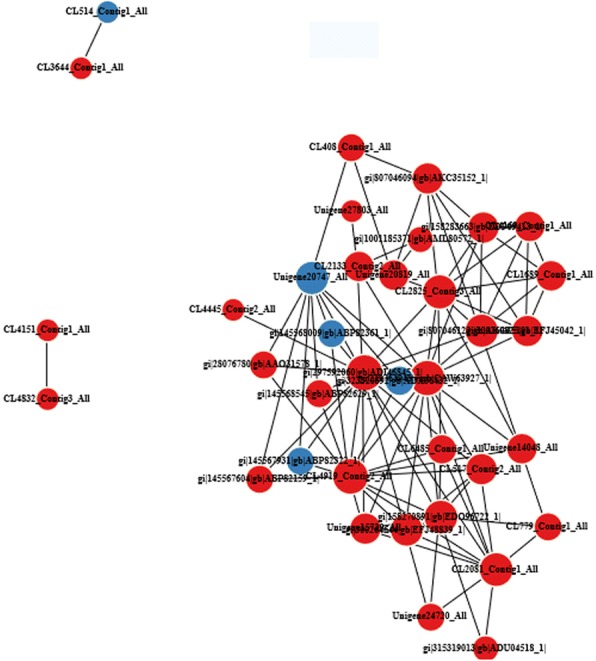
Methods: In this study, isobaric tags for relative and absolute quantitation (iTRAQ) labelling was applied in combination with the liquid chromatography-tandem mass spectrometry (LC-MS/MS) to conduct comparative proteomic analysis of the response of U. prolifera to high-temperature stress and to elucidate the involvement of this response in adaptation mechanisms. Differentially expressed proteins (DEPs) of U. prolifera under high temperature (denote UpHT) compared with the control (UpC) were identified. Bioinformatic analyses including GO analysis, pathway analysis, and pathway enrichment analysis was performed to analyse the key metabolic pathways that underlie the thermal tolerance mechanism through protein networks. Quantitative real-time PCR and western blot were performed to validate selected proteins.
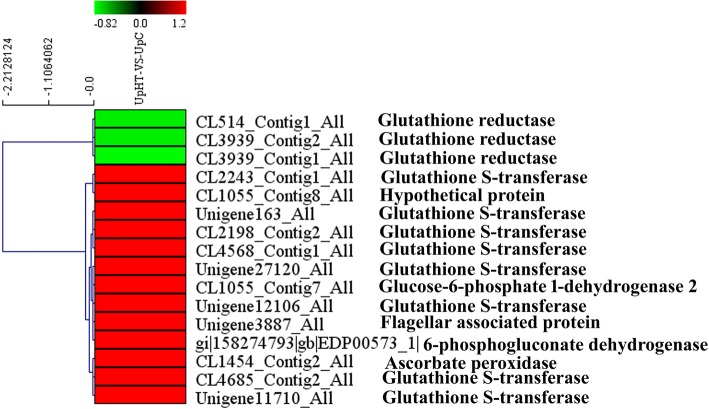
Results: In the present study, 1223 DEPs were identified under high temperature compared with the control, which included 790 up-regulated and 433 down-regulated proteins. The high-temperature stimulus mainly induced the expression of glutathione S-transferase, heat shock protein, ascorbate peroxidase, manganese superoxide dismutase, ubiquitin-related protein, lhcSR, rubisco activase, serine/threonine protein kinase 2, adenylate kinase, Ca2+-dependent protein kinase (CDPK), disease resistance protein EDS1, metacaspase type II, NDPK2a, 26S proteasome regulatory subunit, ubiquinone oxidoreductase, ATP synthase subunit, SnRK2s, and cytochrome P450. The down-regulated proteins were photosynthesis-related proteins, glutathione reductase, catalase-peroxidase, thioredoxin, thioredoxin peroxidase, PP2C, and carbon fixation-related proteins. Furthermore, biological index analysis indicated that protein content and SOD activity decreased; the value of Fv/Fm dropped to the lowest point after culture for 96 h. However, APX activity and MDA content increased under high temperature.
Conclusion: The present study implied an increase in proteins that were associated with the stress response, oxidative phosphorylation, the cytokinin signal transduction pathway, the abscisic acid signal transduction pathway, and the glutathione metabolism pathway. Proteins that were associated with photosynthesis, carbon fixation in photosynthesis organisms, and the photosynthesis antenna protein pathway were decreased. These pathways played a pivotal role in high temperature regulation. These novel proteins provide a good starting point for further research into their functions using genetic or other approaches. These findings significantly improve the understanding of the molecular mechanisms involved in the tolerance of algae to high-temperature stress.
期刊介绍:
Proteome Science is an open access journal publishing research in the area of systems studies. Proteome Science considers manuscripts based on all aspects of functional and structural proteomics, genomics, metabolomics, systems analysis and metabiome analysis. It encourages the submissions of studies that use large-scale or systems analysis of biomolecules in a cellular, organismal and/or environmental context.
Studies that describe novel biological or clinical insights as well as methods-focused studies that describe novel methods for the large-scale study of any and all biomolecules in cells and tissues, such as mass spectrometry, protein and nucleic acid microarrays, genomics, next-generation sequencing and computational algorithms and methods are all within the scope of Proteome Science, as are electron topography, structural methods, proteogenomics, chemical proteomics, stem cell proteomics, organelle proteomics, plant and microbial proteomics.
In spite of its name, Proteome Science considers all aspects of large-scale and systems studies because ultimately any mechanism that results in genomic and metabolomic changes will affect or be affected by the proteome. To reflect this intrinsic relationship of biological systems, Proteome Science will consider all such articles.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
