{"title":"通过全外显子组测序在韩国2名患有Alport综合征的女孩中发现COL4A5从头突变。","authors":"Kyoung Hee Han, Jong Eun Park, Chang-Seok Ki","doi":"10.3345/kjp.2018.06772","DOIUrl":null,"url":null,"abstract":"<p><p>Alport syndrome (ATS) is an inherited glomerular disease caused by mutations in one of the type IV collagen novel chains (α3, α4, and α5). ATS is characterized by persistent microscopic hematuria that starts during infancy, eventually leading to either progressive nephritis or end-stage renal disease. There are 3 known genetic forms of ATS, namely X-linked ATS, autosomal recessive ATS, and autosomal dominant ATS. About 80% of patients with ATS have X-linked ATS, which is caused by mutations in the type IV collagen α5 chain gene, COL4A5. Although an 80% mutation detection rate is observed in men with X-linked ATS, some difficulties do exist in the genetic diagnosis of ATS. Most mutations are point mutations without hotspots in the COL4A3, COL4A4, and COL4A5 genes. Further, there are insufficient data on the detection of COL4A3 and COL4A4 mutations for their comparison between patients with autosomal recessive or dominant ATS. Therefore, diagnosis of ATS in female patients with no apparent family history can be challenging. Therefore, in this study, we used whole-exome sequencing (WES) to identify mutations in type IV collagen in 2 girls with glomerular basement membrane structural changes suspected to be associated with ATS; these patients had no relevant family history. Our results revealed de novo c.4688G>A (p.Arg1563Gln) and c.2714G>A (p.Gly905Asp) mutations in COL4A5. Therefore, we suggest that WES is an effective approach to obtain genetic information in ATS, particularly in female patients without a relevant family history, to detect unexpected DNA variations.</p>","PeriodicalId":17863,"journal":{"name":"Korean Journal of Pediatrics","volume":"62 5","pages":"193-197"},"PeriodicalIF":0.0000,"publicationDate":"2019-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/88/e8/kjp-2018-06772.PMC6528060.pdf","citationCount":"6","resultStr":"{\"title\":\"De novo mutations in COL4A5 identified by whole exome sequencing in 2 girls with Alport syndrome in Korea.\",\"authors\":\"Kyoung Hee Han, Jong Eun Park, Chang-Seok Ki\",\"doi\":\"10.3345/kjp.2018.06772\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Alport syndrome (ATS) is an inherited glomerular disease caused by mutations in one of the type IV collagen novel chains (α3, α4, and α5). ATS is characterized by persistent microscopic hematuria that starts during infancy, eventually leading to either progressive nephritis or end-stage renal disease. There are 3 known genetic forms of ATS, namely X-linked ATS, autosomal recessive ATS, and autosomal dominant ATS. About 80% of patients with ATS have X-linked ATS, which is caused by mutations in the type IV collagen α5 chain gene, COL4A5. Although an 80% mutation detection rate is observed in men with X-linked ATS, some difficulties do exist in the genetic diagnosis of ATS. Most mutations are point mutations without hotspots in the COL4A3, COL4A4, and COL4A5 genes. Further, there are insufficient data on the detection of COL4A3 and COL4A4 mutations for their comparison between patients with autosomal recessive or dominant ATS. Therefore, diagnosis of ATS in female patients with no apparent family history can be challenging. Therefore, in this study, we used whole-exome sequencing (WES) to identify mutations in type IV collagen in 2 girls with glomerular basement membrane structural changes suspected to be associated with ATS; these patients had no relevant family history. Our results revealed de novo c.4688G>A (p.Arg1563Gln) and c.2714G>A (p.Gly905Asp) mutations in COL4A5. Therefore, we suggest that WES is an effective approach to obtain genetic information in ATS, particularly in female patients without a relevant family history, to detect unexpected DNA variations.</p>\",\"PeriodicalId\":17863,\"journal\":{\"name\":\"Korean Journal of Pediatrics\",\"volume\":\"62 5\",\"pages\":\"193-197\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2019-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/88/e8/kjp-2018-06772.PMC6528060.pdf\",\"citationCount\":\"6\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Korean Journal of Pediatrics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3345/kjp.2018.06772\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2018/11/26 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Korean Journal of Pediatrics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3345/kjp.2018.06772","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/11/26 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
De novo mutations in COL4A5 identified by whole exome sequencing in 2 girls with Alport syndrome in Korea.
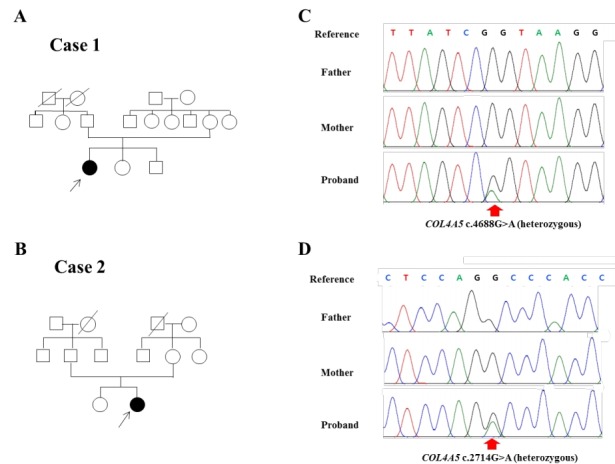
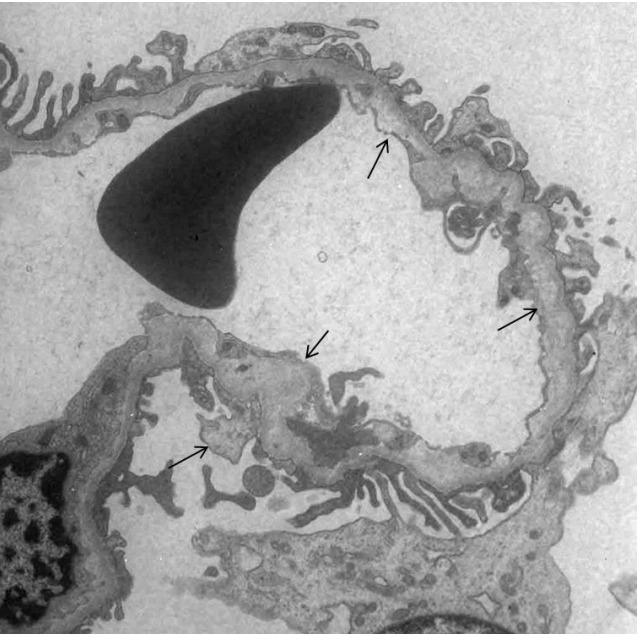
Alport syndrome (ATS) is an inherited glomerular disease caused by mutations in one of the type IV collagen novel chains (α3, α4, and α5). ATS is characterized by persistent microscopic hematuria that starts during infancy, eventually leading to either progressive nephritis or end-stage renal disease. There are 3 known genetic forms of ATS, namely X-linked ATS, autosomal recessive ATS, and autosomal dominant ATS. About 80% of patients with ATS have X-linked ATS, which is caused by mutations in the type IV collagen α5 chain gene, COL4A5. Although an 80% mutation detection rate is observed in men with X-linked ATS, some difficulties do exist in the genetic diagnosis of ATS. Most mutations are point mutations without hotspots in the COL4A3, COL4A4, and COL4A5 genes. Further, there are insufficient data on the detection of COL4A3 and COL4A4 mutations for their comparison between patients with autosomal recessive or dominant ATS. Therefore, diagnosis of ATS in female patients with no apparent family history can be challenging. Therefore, in this study, we used whole-exome sequencing (WES) to identify mutations in type IV collagen in 2 girls with glomerular basement membrane structural changes suspected to be associated with ATS; these patients had no relevant family history. Our results revealed de novo c.4688G>A (p.Arg1563Gln) and c.2714G>A (p.Gly905Asp) mutations in COL4A5. Therefore, we suggest that WES is an effective approach to obtain genetic information in ATS, particularly in female patients without a relevant family history, to detect unexpected DNA variations.
期刊介绍:
Korean J Pediatr covers clinical and research works relevant to all aspects of child healthcare. The journal aims to serve pediatricians through the prompt publication of significant advances in any field of pediatrics and to rapidly disseminate recently updated knowledge to the public. Additionally, it will initiate dynamic, international, academic discussions concerning the major topics related to pediatrics. Manuscripts are categorized as review articles, original articles, and case reports. Areas of specific interest include: Growth and development, Neonatology, Pediatric neurology, Pediatric nephrology, Pediatric endocrinology, Pediatric cardiology, Pediatric allergy, Pediatric pulmonology, Pediatric infectious diseases, Pediatric immunology, Pediatric hemato-oncology, Pediatric gastroenterology, Nutrition, Human genetics, Metabolic diseases, Adolescence medicine, General pediatrics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
