{"title":"神经性疼痛的t型通道——恶棍还是受害者?","authors":"Norbert Weiss","doi":"10.1080/19336950.2020.1740487","DOIUrl":null,"url":null,"abstract":"Neuropathic pain syndromes affect between 30 and 50% of the world population and represent a significant burden for patients, society, and healthcare systems. Many hypotheses have been formulated about the mechanisms of neuropathic pain among which elevated expression of T-type calcium channels in peripheral nociceptive nerve fibers (so-called “nociceptors”) is seen as a hallmark in several experimental pain models [1]. Nociceptors have their cell bodies in the dorsal root ganglia (DRG) and express predominantly the Cav3.2 channel subtype whose primary function is to regulate neuronal firing and synaptic transmission at dorsal horn synapses [2]. Given these important functions in peripheral sensory neurons, aberrant expression of T-type channels in primary pain fibers comes as a pertinent cellular mechanism of neuropathic pain syndromes. How this up-regulation of T-type channels occurs at a mechanistic level has been the subject of a great deal of research in recent years and several studies pointed to a role of post-translationalmodification of the channel protein. Post-translational modification refers to changes a protein may undergo after translation (cleavage and/or covalent addition of chemical moieties) and serves as a secondary level of control to fine tune its functional expression. While post-translational modification of proteins is an essential part of cellular homeostasis, it has become increasingly evident that this process is altered in pathological conditions including pain syndromes. Using a mouse model or peripheral nerve injury-induced neuropathic pain, Garcia-Caballero et al., reported a decreased ubiquitinylation of Cav3.2 channels in primary afferent nerve fibers [3]. Biochemical analysis revealed that this effect was mediated by the up-regulation of the deubiquitinylating enzyme USP5 resulting in the accumulation of Cav3.2 in the plasma membrane. Importantly, the authors showed that prophylactic knockdown of USP5, or prophylactic disruption of the Cav3.2/USP5 complex, was sufficient to prevent nerve-injury-induced mechanical and thermal hyperalgesia demonstrating the causal implication of the ubiquitinylation machinery in the development of neuropathic pain in this experimental model. In yet another study using the same experimental pain model, the authors reported a decreased SUMOylation of USP5 in peripheral nociceptive nerve fibers [4]. Given that SUMOylation of USP5 negatively regulates its ability to interact with Cav3.2, decreased SUMOylated USP5 during nerve injury would favor Cav3.2/USP5 interaction. This would add to the already elevated level of USP5, which would enhance the deubiquitinylation of Cav3.2 and further potentiate the expression of the channel in the plasma membrane. Asparagine (N)-linked glycosylation is another type of post-translational modification that has been reported to potentially contribute to peripheral painful diabetic neuropathy. Several in vitro studies have documented the functional importance of N-glycosylation for Cav3.2 channels [5–7]. Of particular relevance for painful diabetic neuropathy is the observation that surface expression of recombinant Cav3.2 is enhanced in cells exposed to elevated glucose levels and this phenomenon requires the glycosylation of the channel at asparagines N192 and N1466. Along these lines, Orestes et al., demonstrated that in vitro removal of sialic acid moieties from complex glycan structures using neuraminidase normalized T-type currents in DRG neurons from ob/ob mice (a","PeriodicalId":72555,"journal":{"name":"Channels (Austin, Tex.)","volume":" ","pages":"98-100"},"PeriodicalIF":3.2000,"publicationDate":"2020-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1080/19336950.2020.1740487","citationCount":"2","resultStr":"{\"title\":\"T-type channels in neuropathic pain - Villain or victim?\",\"authors\":\"Norbert Weiss\",\"doi\":\"10.1080/19336950.2020.1740487\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Neuropathic pain syndromes affect between 30 and 50% of the world population and represent a significant burden for patients, society, and healthcare systems. Many hypotheses have been formulated about the mechanisms of neuropathic pain among which elevated expression of T-type calcium channels in peripheral nociceptive nerve fibers (so-called “nociceptors”) is seen as a hallmark in several experimental pain models [1]. Nociceptors have their cell bodies in the dorsal root ganglia (DRG) and express predominantly the Cav3.2 channel subtype whose primary function is to regulate neuronal firing and synaptic transmission at dorsal horn synapses [2]. Given these important functions in peripheral sensory neurons, aberrant expression of T-type channels in primary pain fibers comes as a pertinent cellular mechanism of neuropathic pain syndromes. How this up-regulation of T-type channels occurs at a mechanistic level has been the subject of a great deal of research in recent years and several studies pointed to a role of post-translationalmodification of the channel protein. Post-translational modification refers to changes a protein may undergo after translation (cleavage and/or covalent addition of chemical moieties) and serves as a secondary level of control to fine tune its functional expression. While post-translational modification of proteins is an essential part of cellular homeostasis, it has become increasingly evident that this process is altered in pathological conditions including pain syndromes. Using a mouse model or peripheral nerve injury-induced neuropathic pain, Garcia-Caballero et al., reported a decreased ubiquitinylation of Cav3.2 channels in primary afferent nerve fibers [3]. Biochemical analysis revealed that this effect was mediated by the up-regulation of the deubiquitinylating enzyme USP5 resulting in the accumulation of Cav3.2 in the plasma membrane. Importantly, the authors showed that prophylactic knockdown of USP5, or prophylactic disruption of the Cav3.2/USP5 complex, was sufficient to prevent nerve-injury-induced mechanical and thermal hyperalgesia demonstrating the causal implication of the ubiquitinylation machinery in the development of neuropathic pain in this experimental model. In yet another study using the same experimental pain model, the authors reported a decreased SUMOylation of USP5 in peripheral nociceptive nerve fibers [4]. Given that SUMOylation of USP5 negatively regulates its ability to interact with Cav3.2, decreased SUMOylated USP5 during nerve injury would favor Cav3.2/USP5 interaction. This would add to the already elevated level of USP5, which would enhance the deubiquitinylation of Cav3.2 and further potentiate the expression of the channel in the plasma membrane. Asparagine (N)-linked glycosylation is another type of post-translational modification that has been reported to potentially contribute to peripheral painful diabetic neuropathy. Several in vitro studies have documented the functional importance of N-glycosylation for Cav3.2 channels [5–7]. Of particular relevance for painful diabetic neuropathy is the observation that surface expression of recombinant Cav3.2 is enhanced in cells exposed to elevated glucose levels and this phenomenon requires the glycosylation of the channel at asparagines N192 and N1466. Along these lines, Orestes et al., demonstrated that in vitro removal of sialic acid moieties from complex glycan structures using neuraminidase normalized T-type currents in DRG neurons from ob/ob mice (a\",\"PeriodicalId\":72555,\"journal\":{\"name\":\"Channels (Austin, Tex.)\",\"volume\":\" \",\"pages\":\"98-100\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2020-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1080/19336950.2020.1740487\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Channels (Austin, Tex.)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1080/19336950.2020.1740487\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Channels (Austin, Tex.)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1080/19336950.2020.1740487","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
T-type channels in neuropathic pain - Villain or victim?
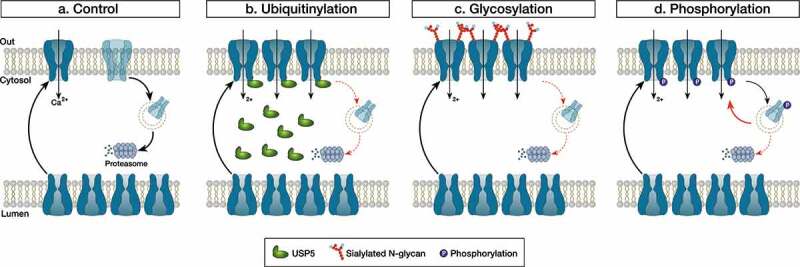
Neuropathic pain syndromes affect between 30 and 50% of the world population and represent a significant burden for patients, society, and healthcare systems. Many hypotheses have been formulated about the mechanisms of neuropathic pain among which elevated expression of T-type calcium channels in peripheral nociceptive nerve fibers (so-called “nociceptors”) is seen as a hallmark in several experimental pain models [1]. Nociceptors have their cell bodies in the dorsal root ganglia (DRG) and express predominantly the Cav3.2 channel subtype whose primary function is to regulate neuronal firing and synaptic transmission at dorsal horn synapses [2]. Given these important functions in peripheral sensory neurons, aberrant expression of T-type channels in primary pain fibers comes as a pertinent cellular mechanism of neuropathic pain syndromes. How this up-regulation of T-type channels occurs at a mechanistic level has been the subject of a great deal of research in recent years and several studies pointed to a role of post-translationalmodification of the channel protein. Post-translational modification refers to changes a protein may undergo after translation (cleavage and/or covalent addition of chemical moieties) and serves as a secondary level of control to fine tune its functional expression. While post-translational modification of proteins is an essential part of cellular homeostasis, it has become increasingly evident that this process is altered in pathological conditions including pain syndromes. Using a mouse model or peripheral nerve injury-induced neuropathic pain, Garcia-Caballero et al., reported a decreased ubiquitinylation of Cav3.2 channels in primary afferent nerve fibers [3]. Biochemical analysis revealed that this effect was mediated by the up-regulation of the deubiquitinylating enzyme USP5 resulting in the accumulation of Cav3.2 in the plasma membrane. Importantly, the authors showed that prophylactic knockdown of USP5, or prophylactic disruption of the Cav3.2/USP5 complex, was sufficient to prevent nerve-injury-induced mechanical and thermal hyperalgesia demonstrating the causal implication of the ubiquitinylation machinery in the development of neuropathic pain in this experimental model. In yet another study using the same experimental pain model, the authors reported a decreased SUMOylation of USP5 in peripheral nociceptive nerve fibers [4]. Given that SUMOylation of USP5 negatively regulates its ability to interact with Cav3.2, decreased SUMOylated USP5 during nerve injury would favor Cav3.2/USP5 interaction. This would add to the already elevated level of USP5, which would enhance the deubiquitinylation of Cav3.2 and further potentiate the expression of the channel in the plasma membrane. Asparagine (N)-linked glycosylation is another type of post-translational modification that has been reported to potentially contribute to peripheral painful diabetic neuropathy. Several in vitro studies have documented the functional importance of N-glycosylation for Cav3.2 channels [5–7]. Of particular relevance for painful diabetic neuropathy is the observation that surface expression of recombinant Cav3.2 is enhanced in cells exposed to elevated glucose levels and this phenomenon requires the glycosylation of the channel at asparagines N192 and N1466. Along these lines, Orestes et al., demonstrated that in vitro removal of sialic acid moieties from complex glycan structures using neuraminidase normalized T-type currents in DRG neurons from ob/ob mice (a


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
