Srdjan Pusara, Wolfgang Wenzel and Mariana Kozlowska
{"title":"用混合泊松-玻尔兹曼和扩展DLVO理论精确计算蛋白质的二次渗透病毒系数","authors":"Srdjan Pusara, Wolfgang Wenzel and Mariana Kozlowska","doi":"10.1039/D3ME00086A","DOIUrl":null,"url":null,"abstract":"<p >The state of proteins in aqueous solution is determined by weak, nonspecific interactions affected by pH, solvent composition, and ionic strength. Protein–protein interactions play a crucial role in determining protein stability and solubility. The second osmotic coefficient (<em>B</em><small><sub>22</sub></small>) provides insight into effective interactions between proteins in solution. Models for calculating <em>B</em><small><sub>22</sub></small> are valuable for estimating interactions, explaining measured phenomena, and reducing experimental time. However, existing models, like the Derjaguin–Landau–Verwey–Overbeek (DLVO) theory, assume a simple spherical shape for proteins. Owing to the fact that proteins exhibit diverse shapes and charge distributions, influencing their electrostatic properties and overall interactions, DLVO accuracy is significantly reduced for nonspherical proteins. To address this limitation, we introduce the xDLVO-CGhybr model, which combines Poisson–Boltzmann (PB) and Debye–Hückel (DH) theories to account for electrostatic interactions between proteins. PB is used for short intermolecular distances (<2 nm) with an all-atom resolution, while DH is employed for longer distances on a coarse-grained level. Additionally, xDLVO-CGhybr incorporates an improved coarse-grained Lennard-Jones (LJ) potential derived directly from the all-atom potential to capture dispersion interactions. This model improves the calculated <em>B</em><small><sub>22</sub></small> values compared to existing models and can be applied to proteins with arbitrary shape and charge under various solvent conditions (up to 1 M monovalent salt concentration). We demonstrate the application of xDLVO-CGhybr to bovine trypsin inhibitor, ribonuclease A, chymotrypsinogen, concanavalin A, bovine serum albumin, and human immunoglobulin type I proteins, validating the model against experimental data.</p>","PeriodicalId":91,"journal":{"name":"Molecular Systems Design & Engineering","volume":" 9","pages":" 1203-1219"},"PeriodicalIF":3.2000,"publicationDate":"2023-07-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2023/me/d3me00086a?page=search","citationCount":"0","resultStr":"{\"title\":\"Accurate calculation of second osmotic virial coefficients of proteins using mixed Poisson–Boltzmann and extended DLVO theory†\",\"authors\":\"Srdjan Pusara, Wolfgang Wenzel and Mariana Kozlowska\",\"doi\":\"10.1039/D3ME00086A\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The state of proteins in aqueous solution is determined by weak, nonspecific interactions affected by pH, solvent composition, and ionic strength. Protein–protein interactions play a crucial role in determining protein stability and solubility. The second osmotic coefficient (<em>B</em><small><sub>22</sub></small>) provides insight into effective interactions between proteins in solution. Models for calculating <em>B</em><small><sub>22</sub></small> are valuable for estimating interactions, explaining measured phenomena, and reducing experimental time. However, existing models, like the Derjaguin–Landau–Verwey–Overbeek (DLVO) theory, assume a simple spherical shape for proteins. Owing to the fact that proteins exhibit diverse shapes and charge distributions, influencing their electrostatic properties and overall interactions, DLVO accuracy is significantly reduced for nonspherical proteins. To address this limitation, we introduce the xDLVO-CGhybr model, which combines Poisson–Boltzmann (PB) and Debye–Hückel (DH) theories to account for electrostatic interactions between proteins. PB is used for short intermolecular distances (<2 nm) with an all-atom resolution, while DH is employed for longer distances on a coarse-grained level. Additionally, xDLVO-CGhybr incorporates an improved coarse-grained Lennard-Jones (LJ) potential derived directly from the all-atom potential to capture dispersion interactions. This model improves the calculated <em>B</em><small><sub>22</sub></small> values compared to existing models and can be applied to proteins with arbitrary shape and charge under various solvent conditions (up to 1 M monovalent salt concentration). We demonstrate the application of xDLVO-CGhybr to bovine trypsin inhibitor, ribonuclease A, chymotrypsinogen, concanavalin A, bovine serum albumin, and human immunoglobulin type I proteins, validating the model against experimental data.</p>\",\"PeriodicalId\":91,\"journal\":{\"name\":\"Molecular Systems Design & Engineering\",\"volume\":\" 9\",\"pages\":\" 1203-1219\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2023-07-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2023/me/d3me00086a?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Systems Design & Engineering\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2023/me/d3me00086a\",\"RegionNum\":3,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Systems Design & Engineering","FirstCategoryId":"5","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2023/me/d3me00086a","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
蛋白质在水溶液中的状态是由受pH值、溶剂组成和离子强度影响的弱、非特异性相互作用决定的。蛋白质之间的相互作用在决定蛋白质的稳定性和溶解度方面起着至关重要的作用。第二渗透系数(B22)提供了对溶液中蛋白质之间有效相互作用的洞察。计算B22的模型对于估计相互作用、解释测量现象和减少实验时间是有价值的。然而,现有的模型,如Derjaguin-Landau-Verwey-Overbeek (DLVO)理论,假设蛋白质是一个简单的球形。由于蛋白质具有不同的形状和电荷分布,这影响了它们的静电特性和整体相互作用,因此对于非球形蛋白质,DLVO精度显着降低。为了解决这一限制,我们引入了xDLVO-CGhybr模型,该模型结合了泊松-玻尔兹曼(PB)和德拜- h ckel (DH)理论来解释蛋白质之间的静电相互作用。PB用于全原子分辨率的短分子间距离(2nm),而DH用于较长距离的粗粒度水平。此外,xDLVO-CGhybr结合了一个改进的粗粒度Lennard-Jones (LJ)势,直接来自全原子势,以捕获色散相互作用。与现有模型相比,该模型改进了计算的B22值,并可应用于各种溶剂条件下(高达1 M单价盐浓度)具有任意形状和电荷的蛋白质。我们展示了xDLVO-CGhybr在牛胰蛋白酶抑制剂、核糖核酸酶A、糜凝胰蛋白酶原、豆豆蛋白A、牛血清白蛋白和人免疫球蛋白I型蛋白上的应用,并根据实验数据验证了该模型。
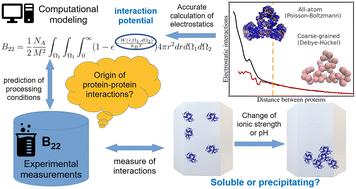
Accurate calculation of second osmotic virial coefficients of proteins using mixed Poisson–Boltzmann and extended DLVO theory†
The state of proteins in aqueous solution is determined by weak, nonspecific interactions affected by pH, solvent composition, and ionic strength. Protein–protein interactions play a crucial role in determining protein stability and solubility. The second osmotic coefficient (B22) provides insight into effective interactions between proteins in solution. Models for calculating B22 are valuable for estimating interactions, explaining measured phenomena, and reducing experimental time. However, existing models, like the Derjaguin–Landau–Verwey–Overbeek (DLVO) theory, assume a simple spherical shape for proteins. Owing to the fact that proteins exhibit diverse shapes and charge distributions, influencing their electrostatic properties and overall interactions, DLVO accuracy is significantly reduced for nonspherical proteins. To address this limitation, we introduce the xDLVO-CGhybr model, which combines Poisson–Boltzmann (PB) and Debye–Hückel (DH) theories to account for electrostatic interactions between proteins. PB is used for short intermolecular distances (<2 nm) with an all-atom resolution, while DH is employed for longer distances on a coarse-grained level. Additionally, xDLVO-CGhybr incorporates an improved coarse-grained Lennard-Jones (LJ) potential derived directly from the all-atom potential to capture dispersion interactions. This model improves the calculated B22 values compared to existing models and can be applied to proteins with arbitrary shape and charge under various solvent conditions (up to 1 M monovalent salt concentration). We demonstrate the application of xDLVO-CGhybr to bovine trypsin inhibitor, ribonuclease A, chymotrypsinogen, concanavalin A, bovine serum albumin, and human immunoglobulin type I proteins, validating the model against experimental data.
期刊介绍:
Molecular Systems Design & Engineering provides a hub for cutting-edge research into how understanding of molecular properties, behaviour and interactions can be used to design and assemble better materials, systems, and processes to achieve specific functions. These may have applications of technological significance and help address global challenges.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
