下载PDF
{"title":"组蛋白纯化结合高分辨率质谱法检测秀丽隐杆线虫组蛋白翻译后修饰和组蛋白变异","authors":"Lluís Millan-Ariño, Zuo-Fei Yuan, Marlies E. Oomen, Simone Brandenburg, Alexey Chernobrovkin, Jérôme Salignon, Lioba Körner, Roman A. Zubarev, Benjamin A. Garcia, Christian G. Riedel","doi":"10.1002/cpps.114","DOIUrl":null,"url":null,"abstract":"Histones are the major proteinaceous component of chromatin in eukaryotic cells and an important part of the epigenome, affecting most DNA‐related events, including transcription, DNA replication, and chromosome segregation. The properties of histones are greatly influenced by their post‐translational modifications (PTMs), over 200 of which are known today. Given this large number, researchers need sophisticated methods to study histone PTMs comprehensively. In particular, mass spectrometry (MS)−based approaches have gained popularity, allowing for the quantification of dozens of histone PTMs at once. Using these approaches, even the study of co‐occurring PTMs and the discovery of novel PTMs become feasible. The success of MS‐based approaches relies substantially on obtaining pure and well‐preserved histones for analysis, which can be difficult depending on the source material. Caenorhabditis elegans has been a popular model organism to study the epigenome, but isolation of pure histones from these animals has been challenging. Here, we address this issue, presenting a method for efficient isolation of pure histone proteins from C. elegans at good yield. Further, we describe an MS pipeline optimized for accurate relative quantification of histone PTMs from C. elegans. We alkylate and tryptically digest the histones, analyze them by bottom‐up MS, and then evaluate the resulting data by a C. elegans−adapted version of the software EpiProfile 2.0. Finally, we show the utility of this pipeline by determining differences in histone PTMs between C. elegans strains that age at different rates and thereby achieve very different lifespans. © 2020 The Authors.","PeriodicalId":10866,"journal":{"name":"Current Protocols in Protein Science","volume":"102 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2020-09-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1002/cpps.114","citationCount":"4","resultStr":"{\"title\":\"Histone Purification Combined with High-Resolution Mass Spectrometry to Examine Histone Post-Translational Modifications and Histone Variants in Caenorhabditis elegans\",\"authors\":\"Lluís Millan-Ariño, Zuo-Fei Yuan, Marlies E. Oomen, Simone Brandenburg, Alexey Chernobrovkin, Jérôme Salignon, Lioba Körner, Roman A. Zubarev, Benjamin A. Garcia, Christian G. Riedel\",\"doi\":\"10.1002/cpps.114\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Histones are the major proteinaceous component of chromatin in eukaryotic cells and an important part of the epigenome, affecting most DNA‐related events, including transcription, DNA replication, and chromosome segregation. The properties of histones are greatly influenced by their post‐translational modifications (PTMs), over 200 of which are known today. Given this large number, researchers need sophisticated methods to study histone PTMs comprehensively. In particular, mass spectrometry (MS)−based approaches have gained popularity, allowing for the quantification of dozens of histone PTMs at once. Using these approaches, even the study of co‐occurring PTMs and the discovery of novel PTMs become feasible. The success of MS‐based approaches relies substantially on obtaining pure and well‐preserved histones for analysis, which can be difficult depending on the source material. Caenorhabditis elegans has been a popular model organism to study the epigenome, but isolation of pure histones from these animals has been challenging. Here, we address this issue, presenting a method for efficient isolation of pure histone proteins from C. elegans at good yield. Further, we describe an MS pipeline optimized for accurate relative quantification of histone PTMs from C. elegans. We alkylate and tryptically digest the histones, analyze them by bottom‐up MS, and then evaluate the resulting data by a C. elegans−adapted version of the software EpiProfile 2.0. Finally, we show the utility of this pipeline by determining differences in histone PTMs between C. elegans strains that age at different rates and thereby achieve very different lifespans. © 2020 The Authors.\",\"PeriodicalId\":10866,\"journal\":{\"name\":\"Current Protocols in Protein Science\",\"volume\":\"102 1\",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-09-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1002/cpps.114\",\"citationCount\":\"4\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Current Protocols in Protein Science\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/cpps.114\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current Protocols in Protein Science","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cpps.114","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 4
引用
批量引用
Histone Purification Combined with High-Resolution Mass Spectrometry to Examine Histone Post-Translational Modifications and Histone Variants in Caenorhabditis elegans
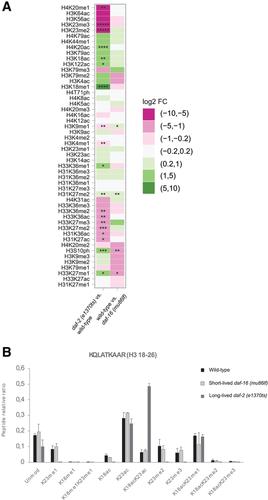
Histones are the major proteinaceous component of chromatin in eukaryotic cells and an important part of the epigenome, affecting most DNA‐related events, including transcription, DNA replication, and chromosome segregation. The properties of histones are greatly influenced by their post‐translational modifications (PTMs), over 200 of which are known today. Given this large number, researchers need sophisticated methods to study histone PTMs comprehensively. In particular, mass spectrometry (MS)−based approaches have gained popularity, allowing for the quantification of dozens of histone PTMs at once. Using these approaches, even the study of co‐occurring PTMs and the discovery of novel PTMs become feasible. The success of MS‐based approaches relies substantially on obtaining pure and well‐preserved histones for analysis, which can be difficult depending on the source material. Caenorhabditis elegans has been a popular model organism to study the epigenome, but isolation of pure histones from these animals has been challenging. Here, we address this issue, presenting a method for efficient isolation of pure histone proteins from C. elegans at good yield. Further, we describe an MS pipeline optimized for accurate relative quantification of histone PTMs from C. elegans. We alkylate and tryptically digest the histones, analyze them by bottom‐up MS, and then evaluate the resulting data by a C. elegans−adapted version of the software EpiProfile 2.0. Finally, we show the utility of this pipeline by determining differences in histone PTMs between C. elegans strains that age at different rates and thereby achieve very different lifespans. © 2020 The Authors.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
