Rong Zhang, Weitao Jiang, Xin Liu, Yanan Duan, Li Xiang, Yanfang Wang, Yuanmao Jiang, Xiang Shen, Xuesen Chen, Chengmiao Yin, Zhiquan Mao
{"title":"基于itraq的moniliforme Fusarium verticillioides对Phloridzin诱导剂响应的定量蛋白质组学分析。","authors":"Rong Zhang, Weitao Jiang, Xin Liu, Yanan Duan, Li Xiang, Yanfang Wang, Yuanmao Jiang, Xiang Shen, Xuesen Chen, Chengmiao Yin, Zhiquan Mao","doi":"10.1186/s12953-021-00170-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Apple replant disease (ARD) has been reported from all major fruit-growing regions of the world, and is often caused by biotic factors (pathogen fungi) and abiotic factors (phenolic compounds). In order to clarify the proteomic differences of Fusarium moniliforme under the action of phloridzin, and to explore the potential mechanism of F. moniliforme as the pathogen of ARD, the role of Fusarium spp. in ARD was further clarified.</p><p><strong>Methods: </strong>In this paper, the quantitative proteomics method iTRAQ analysis technology was used to analyze the proteomic differences of F. moniliforme before and after phloridzin treatment. The differentially expressed protein was validated by qRT-PCR analysis.</p><p><strong>Results: </strong>A total of 4535 proteins were detected, and 293 proteins were found with more than 1.2 times (P< 0.05) differences. In-depth data analysis revealed that 59 proteins were found with more than 1.5 times (P< 0.05) differences, and most proteins were consistent with the result of qRT-PCR. Differentially expressed proteins were influenced a variety of cellular processes, particularly metabolic processes. Among these metabolic pathways, a total of 8 significantly enriched KEGG pathways were identified with at least 2 affiliated proteins with different abundance in conidia and mycelium. Functional pathway analysis indicated that up-regulated proteins were mainly distributed in amino sugar, nucleotide sugar metabolism, glycolysis/ gluconeogenesis and phagosome pathways.</p><p><strong>Conclusions: </strong>This study is the first to perform quantitative proteomic investigation by iTRAQ labeling and LC-MS/MS to identify differentially expressed proteins in F. moniliforme under phloridzin conditions. The results confirmed that F. moniliforme presented a unique protein profile that indicated the adaptive mechanisms of this species to phloridzin environments. The results deepened our understanding of the proteome in F. moniliforme in response to phloridzin inducers and provide a basis for further exploration for improving the efficiency of the fungi as biocontrol agents to control ARD.</p>","PeriodicalId":20857,"journal":{"name":"Proteome Science","volume":"19 1","pages":"2"},"PeriodicalIF":1.6000,"publicationDate":"2021-01-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12953-021-00170-2","citationCount":"4","resultStr":"{\"title\":\"ITRAQ-based quantitative proteomic analysis of Fusarium moniliforme (Fusarium verticillioides) in response to Phloridzin inducers.\",\"authors\":\"Rong Zhang, Weitao Jiang, Xin Liu, Yanan Duan, Li Xiang, Yanfang Wang, Yuanmao Jiang, Xiang Shen, Xuesen Chen, Chengmiao Yin, Zhiquan Mao\",\"doi\":\"10.1186/s12953-021-00170-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Apple replant disease (ARD) has been reported from all major fruit-growing regions of the world, and is often caused by biotic factors (pathogen fungi) and abiotic factors (phenolic compounds). In order to clarify the proteomic differences of Fusarium moniliforme under the action of phloridzin, and to explore the potential mechanism of F. moniliforme as the pathogen of ARD, the role of Fusarium spp. in ARD was further clarified.</p><p><strong>Methods: </strong>In this paper, the quantitative proteomics method iTRAQ analysis technology was used to analyze the proteomic differences of F. moniliforme before and after phloridzin treatment. The differentially expressed protein was validated by qRT-PCR analysis.</p><p><strong>Results: </strong>A total of 4535 proteins were detected, and 293 proteins were found with more than 1.2 times (P< 0.05) differences. In-depth data analysis revealed that 59 proteins were found with more than 1.5 times (P< 0.05) differences, and most proteins were consistent with the result of qRT-PCR. Differentially expressed proteins were influenced a variety of cellular processes, particularly metabolic processes. Among these metabolic pathways, a total of 8 significantly enriched KEGG pathways were identified with at least 2 affiliated proteins with different abundance in conidia and mycelium. Functional pathway analysis indicated that up-regulated proteins were mainly distributed in amino sugar, nucleotide sugar metabolism, glycolysis/ gluconeogenesis and phagosome pathways.</p><p><strong>Conclusions: </strong>This study is the first to perform quantitative proteomic investigation by iTRAQ labeling and LC-MS/MS to identify differentially expressed proteins in F. moniliforme under phloridzin conditions. The results confirmed that F. moniliforme presented a unique protein profile that indicated the adaptive mechanisms of this species to phloridzin environments. The results deepened our understanding of the proteome in F. moniliforme in response to phloridzin inducers and provide a basis for further exploration for improving the efficiency of the fungi as biocontrol agents to control ARD.</p>\",\"PeriodicalId\":20857,\"journal\":{\"name\":\"Proteome Science\",\"volume\":\"19 1\",\"pages\":\"2\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2021-01-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s12953-021-00170-2\",\"citationCount\":\"4\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Proteome Science\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12953-021-00170-2\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteome Science","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12953-021-00170-2","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
ITRAQ-based quantitative proteomic analysis of Fusarium moniliforme (Fusarium verticillioides) in response to Phloridzin inducers.
Background: Apple replant disease (ARD) has been reported from all major fruit-growing regions of the world, and is often caused by biotic factors (pathogen fungi) and abiotic factors (phenolic compounds). In order to clarify the proteomic differences of Fusarium moniliforme under the action of phloridzin, and to explore the potential mechanism of F. moniliforme as the pathogen of ARD, the role of Fusarium spp. in ARD was further clarified.
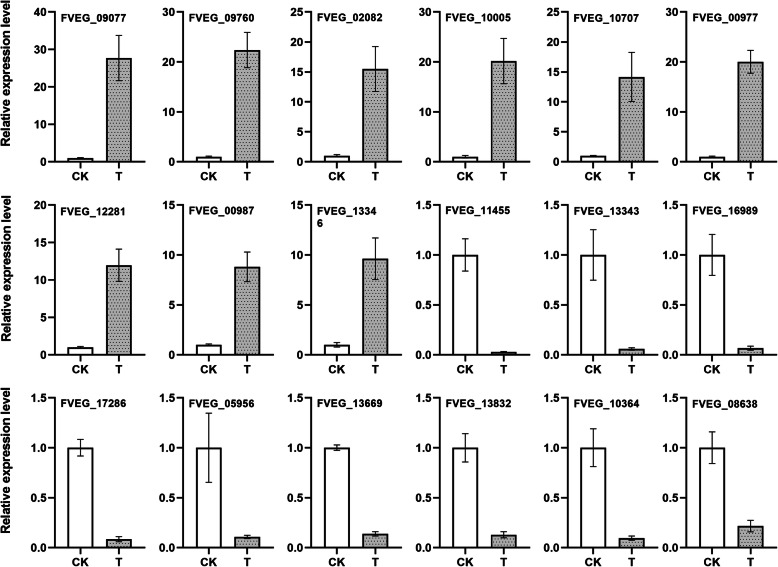
Methods: In this paper, the quantitative proteomics method iTRAQ analysis technology was used to analyze the proteomic differences of F. moniliforme before and after phloridzin treatment. The differentially expressed protein was validated by qRT-PCR analysis.
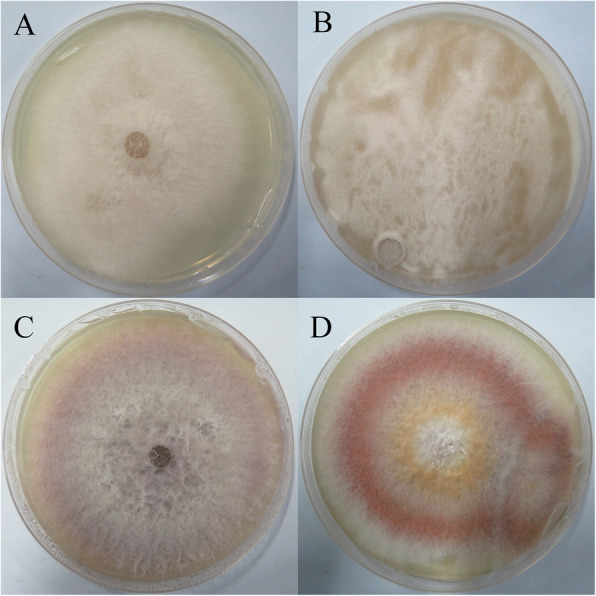
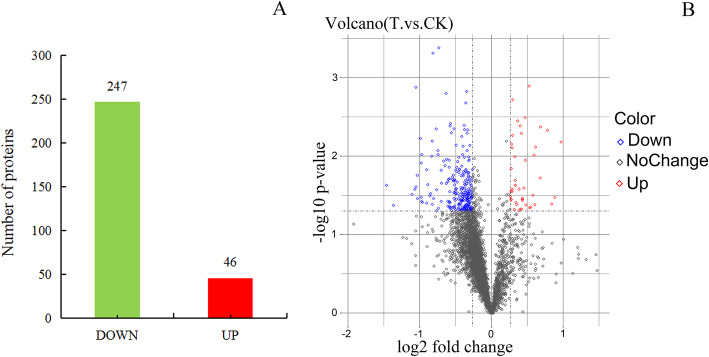
Results: A total of 4535 proteins were detected, and 293 proteins were found with more than 1.2 times (P< 0.05) differences. In-depth data analysis revealed that 59 proteins were found with more than 1.5 times (P< 0.05) differences, and most proteins were consistent with the result of qRT-PCR. Differentially expressed proteins were influenced a variety of cellular processes, particularly metabolic processes. Among these metabolic pathways, a total of 8 significantly enriched KEGG pathways were identified with at least 2 affiliated proteins with different abundance in conidia and mycelium. Functional pathway analysis indicated that up-regulated proteins were mainly distributed in amino sugar, nucleotide sugar metabolism, glycolysis/ gluconeogenesis and phagosome pathways.
Conclusions: This study is the first to perform quantitative proteomic investigation by iTRAQ labeling and LC-MS/MS to identify differentially expressed proteins in F. moniliforme under phloridzin conditions. The results confirmed that F. moniliforme presented a unique protein profile that indicated the adaptive mechanisms of this species to phloridzin environments. The results deepened our understanding of the proteome in F. moniliforme in response to phloridzin inducers and provide a basis for further exploration for improving the efficiency of the fungi as biocontrol agents to control ARD.
期刊介绍:
Proteome Science is an open access journal publishing research in the area of systems studies. Proteome Science considers manuscripts based on all aspects of functional and structural proteomics, genomics, metabolomics, systems analysis and metabiome analysis. It encourages the submissions of studies that use large-scale or systems analysis of biomolecules in a cellular, organismal and/or environmental context.
Studies that describe novel biological or clinical insights as well as methods-focused studies that describe novel methods for the large-scale study of any and all biomolecules in cells and tissues, such as mass spectrometry, protein and nucleic acid microarrays, genomics, next-generation sequencing and computational algorithms and methods are all within the scope of Proteome Science, as are electron topography, structural methods, proteogenomics, chemical proteomics, stem cell proteomics, organelle proteomics, plant and microbial proteomics.
In spite of its name, Proteome Science considers all aspects of large-scale and systems studies because ultimately any mechanism that results in genomic and metabolomic changes will affect or be affected by the proteome. To reflect this intrinsic relationship of biological systems, Proteome Science will consider all such articles.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
