Jiajin Wu, Chao Hou, Yuhao Wang, Zhongyuan Wang, Pu Li, Zengjun Wang
{"title":"透明细胞肾细胞癌中m5C RNA甲基化调控基因的综合分析","authors":"Jiajin Wu, Chao Hou, Yuhao Wang, Zhongyuan Wang, Pu Li, Zengjun Wang","doi":"10.1155/2021/3803724","DOIUrl":null,"url":null,"abstract":"<div>\n <p><i>Background</i>. Recent research found that N5-methylcytosine (m<sup>5</sup>C) was involved in the development and occurrence of numerous cancers. However, the function and mechanism of m<sup>5</sup>C RNA methylation regulators in clear cell renal cell carcinoma (ccRCC) remains undiscovered. This study is aimed at investigating the predictive and clinical value of these m<sup>5</sup>C-related genes in ccRCC. <i>Methods</i>. Based on The Cancer Genome Atlas (TCGA) database, the expression patterns of twelve m<sup>5</sup>C regulators and matched clinicopathological characteristics were downloaded and analyzed. To reveal the relationships between the expression levels of m<sup>5</sup>C-related genes and the prognosis value in ccRCC, consensus clustering analysis was carried out. By univariate Cox analysis and last absolute shrinkage and selection operator (LASSO) Cox regression algorithm, a m<sup>5</sup>C-related risk signature was constructed in the training group and further validated in the testing group and the entire cohort. Then, the predictive ability of survival of this m<sup>5</sup>C-related risk signature was analyzed by Cox regression analysis and nomogram. Functional annotation and single-sample Gene Set Enrichment Analysis (ssGSEA) were applied to further explore the biological function and potential signaling pathways. Furthermore, we performed qRT-PCR experiments and measured global m<sup>5</sup>C RNA methylation level to validate this signature in vitro and tissue samples. <i>Results</i>. In the TCGA-KIRC cohort, we found significant differences in the expression of m<sup>5</sup>C RNA methylation-related genes between ccRCC tissues and normal kidney tissues. Consensus cluster analysis was conducted to separate patients into two m<sup>5</sup>C RNA methylation subtypes. Significantly better outcomes were observed in ccRCC patients in cluster 1 than in cluster 2. m<sup>5</sup>C RNA methylation-related risk score was calculated to evaluate the prognosis of ccRCC patients by seven screened m<sup>5</sup>C RNA methylation regulators (NOP2, NSUN2, NSUN3, NSUN4, NSUN5, TET2, and DNMT3B) in the training cohort. The AUC for the 1-, 2-, and 3-year survival in the training cohort were 0.792, 0.675, and 0.709, respectively, indicating that the risk signature had an excellent prognosis prediction in ccRCC. Additionally, univariate and multivariate Cox regression analyses revealed that the risk signature could be an independent prognostic factor in ccRCC. The results of ssGSEA suggested that the immune cells with different infiltration degrees between the high-risk and low-risk groups were T cells including follicular helper T cells, Th1_cells, Th2_cells, and CD8+_T_cells, and the main differences in immune-related functions between the two groups were the interferon response and T cell costimulation. In addition, qRT-PCR experiments confirmed our results in renal cell lines and tissue samples. <i>Conclusions</i>. According to the seven selected regulatory factors of m<sup>5</sup>C RNA methylation, a risk signature associated with m<sup>5</sup>C methylation that can independently predict prognosis in patients with ccRCC was developed and further verified the predictive efficiency.</p>\n </div>","PeriodicalId":55239,"journal":{"name":"Comparative and Functional Genomics","volume":"2021 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2021-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8497170/pdf/","citationCount":"0","resultStr":"{\"title\":\"Comprehensive Analysis of m5C RNA Methylation Regulator Genes in Clear Cell Renal Cell Carcinoma\",\"authors\":\"Jiajin Wu, Chao Hou, Yuhao Wang, Zhongyuan Wang, Pu Li, Zengjun Wang\",\"doi\":\"10.1155/2021/3803724\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n <p><i>Background</i>. Recent research found that N5-methylcytosine (m<sup>5</sup>C) was involved in the development and occurrence of numerous cancers. However, the function and mechanism of m<sup>5</sup>C RNA methylation regulators in clear cell renal cell carcinoma (ccRCC) remains undiscovered. This study is aimed at investigating the predictive and clinical value of these m<sup>5</sup>C-related genes in ccRCC. <i>Methods</i>. Based on The Cancer Genome Atlas (TCGA) database, the expression patterns of twelve m<sup>5</sup>C regulators and matched clinicopathological characteristics were downloaded and analyzed. To reveal the relationships between the expression levels of m<sup>5</sup>C-related genes and the prognosis value in ccRCC, consensus clustering analysis was carried out. By univariate Cox analysis and last absolute shrinkage and selection operator (LASSO) Cox regression algorithm, a m<sup>5</sup>C-related risk signature was constructed in the training group and further validated in the testing group and the entire cohort. Then, the predictive ability of survival of this m<sup>5</sup>C-related risk signature was analyzed by Cox regression analysis and nomogram. Functional annotation and single-sample Gene Set Enrichment Analysis (ssGSEA) were applied to further explore the biological function and potential signaling pathways. Furthermore, we performed qRT-PCR experiments and measured global m<sup>5</sup>C RNA methylation level to validate this signature in vitro and tissue samples. <i>Results</i>. In the TCGA-KIRC cohort, we found significant differences in the expression of m<sup>5</sup>C RNA methylation-related genes between ccRCC tissues and normal kidney tissues. Consensus cluster analysis was conducted to separate patients into two m<sup>5</sup>C RNA methylation subtypes. Significantly better outcomes were observed in ccRCC patients in cluster 1 than in cluster 2. m<sup>5</sup>C RNA methylation-related risk score was calculated to evaluate the prognosis of ccRCC patients by seven screened m<sup>5</sup>C RNA methylation regulators (NOP2, NSUN2, NSUN3, NSUN4, NSUN5, TET2, and DNMT3B) in the training cohort. The AUC for the 1-, 2-, and 3-year survival in the training cohort were 0.792, 0.675, and 0.709, respectively, indicating that the risk signature had an excellent prognosis prediction in ccRCC. Additionally, univariate and multivariate Cox regression analyses revealed that the risk signature could be an independent prognostic factor in ccRCC. The results of ssGSEA suggested that the immune cells with different infiltration degrees between the high-risk and low-risk groups were T cells including follicular helper T cells, Th1_cells, Th2_cells, and CD8+_T_cells, and the main differences in immune-related functions between the two groups were the interferon response and T cell costimulation. In addition, qRT-PCR experiments confirmed our results in renal cell lines and tissue samples. <i>Conclusions</i>. According to the seven selected regulatory factors of m<sup>5</sup>C RNA methylation, a risk signature associated with m<sup>5</sup>C methylation that can independently predict prognosis in patients with ccRCC was developed and further verified the predictive efficiency.</p>\\n </div>\",\"PeriodicalId\":55239,\"journal\":{\"name\":\"Comparative and Functional Genomics\",\"volume\":\"2021 1\",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-09-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8497170/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Comparative and Functional Genomics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1155/2021/3803724\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Comparative and Functional Genomics","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1155/2021/3803724","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
背景。最近的研究发现,n5 -甲基胞嘧啶(m5C)参与了许多癌症的发展和发生。然而,m5C RNA甲基化调控因子在透明细胞肾细胞癌(ccRCC)中的功能和机制仍未被发现。本研究旨在探讨这些m5c相关基因在ccRCC中的预测和临床价值。方法。基于癌症基因组图谱(The Cancer Genome Atlas, TCGA)数据库,下载12个m5C调节因子的表达模式和匹配的临床病理特征并进行分析。为了揭示m5c相关基因表达水平与ccRCC预后价值之间的关系,我们进行了共识聚类分析。通过单因素Cox分析和LASSO (absolute shrink and selection operator) Cox回归算法,在训练组中构建m5c相关风险签名,并在测试组和整个队列中进一步验证。然后,通过Cox回归分析和nomogram分析m5c相关风险特征对生存的预测能力。应用功能注释和单样本基因集富集分析(ssGSEA)进一步探索其生物学功能和潜在的信号通路。此外,我们进行了qRT-PCR实验,并测量了全球m5C RNA甲基化水平,以在体外和组织样本中验证这一特征。结果。在TCGA-KIRC队列中,我们发现ccRCC组织与正常肾脏组织中m5C RNA甲基化相关基因的表达存在显著差异。采用一致聚类分析将患者分为两个m5C RNA甲基化亚型。第一组ccRCC患者的预后明显好于第二组。计算m5C RNA甲基化相关风险评分,通过筛选的7种m5C RNA甲基化调节因子(NOP2、NSUN2、NSUN3、NSUN4、NSUN5、TET2和DNMT3B)在训练队列中评估ccRCC患者的预后。训练队列1年、2年和3年生存率的AUC分别为0.792、0.675和0.709,表明该风险特征在ccRCC中具有很好的预后预测作用。此外,单因素和多因素Cox回归分析显示,风险标志可能是ccRCC的独立预后因素。ssGSEA结果提示,高危组和低危组浸润程度不同的免疫细胞均为T细胞,包括滤泡辅助性T细胞、Th1_cells、Th2_cells和CD8+_T_cells,两组间免疫相关功能的主要差异在于干扰素应答和T细胞共刺激。此外,qRT-PCR实验在肾细胞系和组织样本中证实了我们的结果。结论。根据筛选出的7个m5C RNA甲基化调控因子,建立了可独立预测ccRCC患者预后的m5C甲基化相关风险标志,并进一步验证了其预测效率。
Comprehensive Analysis of m5C RNA Methylation Regulator Genes in Clear Cell Renal Cell Carcinoma
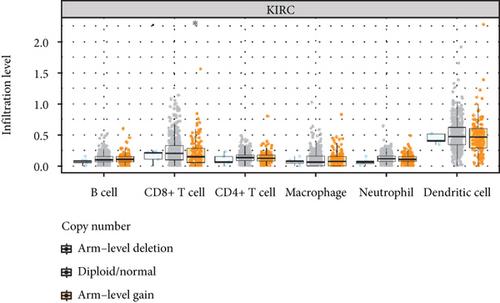
Background. Recent research found that N5-methylcytosine (m5C) was involved in the development and occurrence of numerous cancers. However, the function and mechanism of m5C RNA methylation regulators in clear cell renal cell carcinoma (ccRCC) remains undiscovered. This study is aimed at investigating the predictive and clinical value of these m5C-related genes in ccRCC. Methods. Based on The Cancer Genome Atlas (TCGA) database, the expression patterns of twelve m5C regulators and matched clinicopathological characteristics were downloaded and analyzed. To reveal the relationships between the expression levels of m5C-related genes and the prognosis value in ccRCC, consensus clustering analysis was carried out. By univariate Cox analysis and last absolute shrinkage and selection operator (LASSO) Cox regression algorithm, a m5C-related risk signature was constructed in the training group and further validated in the testing group and the entire cohort. Then, the predictive ability of survival of this m5C-related risk signature was analyzed by Cox regression analysis and nomogram. Functional annotation and single-sample Gene Set Enrichment Analysis (ssGSEA) were applied to further explore the biological function and potential signaling pathways. Furthermore, we performed qRT-PCR experiments and measured global m5C RNA methylation level to validate this signature in vitro and tissue samples. Results. In the TCGA-KIRC cohort, we found significant differences in the expression of m5C RNA methylation-related genes between ccRCC tissues and normal kidney tissues. Consensus cluster analysis was conducted to separate patients into two m5C RNA methylation subtypes. Significantly better outcomes were observed in ccRCC patients in cluster 1 than in cluster 2. m5C RNA methylation-related risk score was calculated to evaluate the prognosis of ccRCC patients by seven screened m5C RNA methylation regulators (NOP2, NSUN2, NSUN3, NSUN4, NSUN5, TET2, and DNMT3B) in the training cohort. The AUC for the 1-, 2-, and 3-year survival in the training cohort were 0.792, 0.675, and 0.709, respectively, indicating that the risk signature had an excellent prognosis prediction in ccRCC. Additionally, univariate and multivariate Cox regression analyses revealed that the risk signature could be an independent prognostic factor in ccRCC. The results of ssGSEA suggested that the immune cells with different infiltration degrees between the high-risk and low-risk groups were T cells including follicular helper T cells, Th1_cells, Th2_cells, and CD8+_T_cells, and the main differences in immune-related functions between the two groups were the interferon response and T cell costimulation. In addition, qRT-PCR experiments confirmed our results in renal cell lines and tissue samples. Conclusions. According to the seven selected regulatory factors of m5C RNA methylation, a risk signature associated with m5C methylation that can independently predict prognosis in patients with ccRCC was developed and further verified the predictive efficiency.


 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
