Juergen R Schaefer, Bilgen Kurt, Alexander Sattler, Günter Klaus, Muhidien Soufi
{"title":"家族性高胆固醇血症的药物遗传学方面,特别关注FHMarburg (FH p.W556R)。","authors":"Juergen R Schaefer, Bilgen Kurt, Alexander Sattler, Günter Klaus, Muhidien Soufi","doi":"10.1007/s11789-012-0041-y","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>Familial hypercholesterolemia (FH) is an autosomal dominant inherited disorder caused by mutations in the low density lipoprotein receptor (LDLR) gene. FH is characterized by elevated plasma LDL cholesterol, premature atherosclerosis, and a high risk of premature myocardial infarction. In general, mutations within LDLR gene can cause five different classes of defects, namely: class I defect: no LDLR synthesis; class II defect: no LDLR transport; class III defect: no low density lipoprotein (LDL) to LDLR binding; class IV defect: no LDLR/LDL internalization; and class V defect: no LDLR recycling. One might expect that both the class of LDLR defect as well as the precise mutation influences the severity of hypercholesterolemia on one hand and the response on drug treatment on the other. To clarify this question we studied the effect of the LDLR mutation p.W556R in two heterozygote subjects.</p><p><strong>Results: </strong>We found that two heterozygote FH patients with the LDLR mutation p.W556R causing a class II LDLR defect (transport defective LDLR) respond exceedingly well to the treatment with simvastatin 40 mg/ezetimibe 10 mg. There was a LDL cholesterol decrease of 55 and 64%, respectively. In contrast, two affected homozygote p.W556R FH patients, in the mean time undergoing LDL apheresis, had no response to statin but a 15% LDL cholesterol decrease on ezetimibe monotherapy.</p><p><strong>Conclusions: </strong>The LDLR mutation p.W556R is a frequent and severe class II defect for FH. The affected homozygote FH patients have a total loss of the functional LDLR and-as expected-do not respond on statin therapy and require LDL apheresis. In contrast, heterozygote FH patients with the same LDLR defect respond exceedingly well to standard lipid-lowering therapy, illustrating that the knowledge of the primary LDLR defect enables us to foresee the expected drug effects.</p>","PeriodicalId":39208,"journal":{"name":"Clinical Research in Cardiology Supplements","volume":" ","pages":"2-6"},"PeriodicalIF":0.0000,"publicationDate":"2012-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1007/s11789-012-0041-y","citationCount":"16","resultStr":"{\"title\":\"Pharmacogenetic aspects in familial hypercholesterolemia with the special focus on FHMarburg (FH p.W556R).\",\"authors\":\"Juergen R Schaefer, Bilgen Kurt, Alexander Sattler, Günter Klaus, Muhidien Soufi\",\"doi\":\"10.1007/s11789-012-0041-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objective: </strong>Familial hypercholesterolemia (FH) is an autosomal dominant inherited disorder caused by mutations in the low density lipoprotein receptor (LDLR) gene. FH is characterized by elevated plasma LDL cholesterol, premature atherosclerosis, and a high risk of premature myocardial infarction. In general, mutations within LDLR gene can cause five different classes of defects, namely: class I defect: no LDLR synthesis; class II defect: no LDLR transport; class III defect: no low density lipoprotein (LDL) to LDLR binding; class IV defect: no LDLR/LDL internalization; and class V defect: no LDLR recycling. One might expect that both the class of LDLR defect as well as the precise mutation influences the severity of hypercholesterolemia on one hand and the response on drug treatment on the other. To clarify this question we studied the effect of the LDLR mutation p.W556R in two heterozygote subjects.</p><p><strong>Results: </strong>We found that two heterozygote FH patients with the LDLR mutation p.W556R causing a class II LDLR defect (transport defective LDLR) respond exceedingly well to the treatment with simvastatin 40 mg/ezetimibe 10 mg. There was a LDL cholesterol decrease of 55 and 64%, respectively. In contrast, two affected homozygote p.W556R FH patients, in the mean time undergoing LDL apheresis, had no response to statin but a 15% LDL cholesterol decrease on ezetimibe monotherapy.</p><p><strong>Conclusions: </strong>The LDLR mutation p.W556R is a frequent and severe class II defect for FH. The affected homozygote FH patients have a total loss of the functional LDLR and-as expected-do not respond on statin therapy and require LDL apheresis. In contrast, heterozygote FH patients with the same LDLR defect respond exceedingly well to standard lipid-lowering therapy, illustrating that the knowledge of the primary LDLR defect enables us to foresee the expected drug effects.</p>\",\"PeriodicalId\":39208,\"journal\":{\"name\":\"Clinical Research in Cardiology Supplements\",\"volume\":\" \",\"pages\":\"2-6\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2012-06-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1007/s11789-012-0041-y\",\"citationCount\":\"16\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Research in Cardiology Supplements\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1007/s11789-012-0041-y\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Research in Cardiology Supplements","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s11789-012-0041-y","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Medicine","Score":null,"Total":0}
Pharmacogenetic aspects in familial hypercholesterolemia with the special focus on FHMarburg (FH p.W556R).
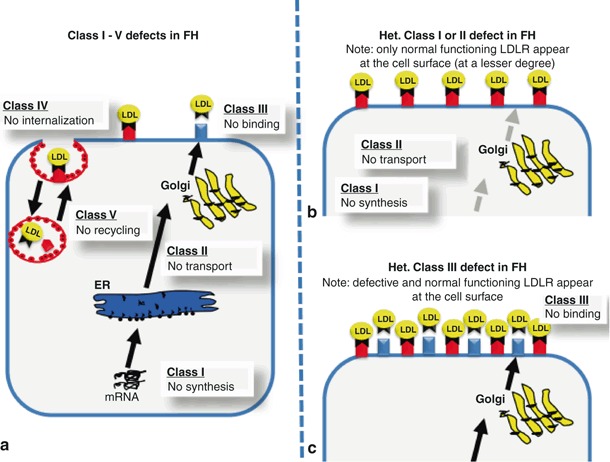
Objective: Familial hypercholesterolemia (FH) is an autosomal dominant inherited disorder caused by mutations in the low density lipoprotein receptor (LDLR) gene. FH is characterized by elevated plasma LDL cholesterol, premature atherosclerosis, and a high risk of premature myocardial infarction. In general, mutations within LDLR gene can cause five different classes of defects, namely: class I defect: no LDLR synthesis; class II defect: no LDLR transport; class III defect: no low density lipoprotein (LDL) to LDLR binding; class IV defect: no LDLR/LDL internalization; and class V defect: no LDLR recycling. One might expect that both the class of LDLR defect as well as the precise mutation influences the severity of hypercholesterolemia on one hand and the response on drug treatment on the other. To clarify this question we studied the effect of the LDLR mutation p.W556R in two heterozygote subjects.
Results: We found that two heterozygote FH patients with the LDLR mutation p.W556R causing a class II LDLR defect (transport defective LDLR) respond exceedingly well to the treatment with simvastatin 40 mg/ezetimibe 10 mg. There was a LDL cholesterol decrease of 55 and 64%, respectively. In contrast, two affected homozygote p.W556R FH patients, in the mean time undergoing LDL apheresis, had no response to statin but a 15% LDL cholesterol decrease on ezetimibe monotherapy.
Conclusions: The LDLR mutation p.W556R is a frequent and severe class II defect for FH. The affected homozygote FH patients have a total loss of the functional LDLR and-as expected-do not respond on statin therapy and require LDL apheresis. In contrast, heterozygote FH patients with the same LDLR defect respond exceedingly well to standard lipid-lowering therapy, illustrating that the knowledge of the primary LDLR defect enables us to foresee the expected drug effects.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
