Marine Peralta, Marie-Christine Combes, Alberto Cenci, Philippe Lashermes, Alexis Dereeper
{"title":"SNiPloid:在全多倍体物种中利用从 RNA-Seq 提取的高通量 SNP 数据的实用工具。","authors":"Marine Peralta, Marie-Christine Combes, Alberto Cenci, Philippe Lashermes, Alexis Dereeper","doi":"10.1155/2013/890123","DOIUrl":null,"url":null,"abstract":"<p><p>High-throughput sequencing is a common approach to discover SNP variants, especially in plant species. However, methods to analyze predicted SNPs are often optimized for diploid plant species whereas many crop species are allopolyploids and combine related but divergent subgenomes (homoeologous chromosome sets). We created a software tool, SNiPloid, that exploits and interprets putative SNPs in the context of allopolyploidy by comparing SNPs from an allopolyploid with those obtained in its modern-day diploid progenitors. SNiPloid can compare SNPs obtained from a sample to estimate the subgenome contribution to the transcriptome or SNPs obtained from two polyploid accessions to search for SNP divergence. </p>","PeriodicalId":73471,"journal":{"name":"International journal of plant genomics","volume":" ","pages":"890123"},"PeriodicalIF":0.0000,"publicationDate":"2013-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3791807/pdf/","citationCount":"0","resultStr":"{\"title\":\"SNiPloid: A Utility to Exploit High-Throughput SNP Data Derived from RNA-Seq in Allopolyploid Species.\",\"authors\":\"Marine Peralta, Marie-Christine Combes, Alberto Cenci, Philippe Lashermes, Alexis Dereeper\",\"doi\":\"10.1155/2013/890123\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>High-throughput sequencing is a common approach to discover SNP variants, especially in plant species. However, methods to analyze predicted SNPs are often optimized for diploid plant species whereas many crop species are allopolyploids and combine related but divergent subgenomes (homoeologous chromosome sets). We created a software tool, SNiPloid, that exploits and interprets putative SNPs in the context of allopolyploidy by comparing SNPs from an allopolyploid with those obtained in its modern-day diploid progenitors. SNiPloid can compare SNPs obtained from a sample to estimate the subgenome contribution to the transcriptome or SNPs obtained from two polyploid accessions to search for SNP divergence. </p>\",\"PeriodicalId\":73471,\"journal\":{\"name\":\"International journal of plant genomics\",\"volume\":\" \",\"pages\":\"890123\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2013-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3791807/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International journal of plant genomics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2013/890123\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2013/9/12 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International journal of plant genomics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2013/890123","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2013/9/12 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
高通量测序是发现 SNP 变异的常用方法,尤其是在植物物种中。然而,分析预测 SNP 的方法通常针对二倍体植物物种进行优化,而许多作物物种是异源多倍体,结合了相关但不同的亚基因组(同源染色体组)。我们开发了一款软件工具 SNiPloid,通过比较来自异源多倍体的 SNPs 和来自其现代二倍体祖先的 SNPs,在异源多倍体的背景下利用和解释推测的 SNPs。SNiPloid 可以比较从一个样本中获得的 SNPs,以估计亚基因组对转录组的贡献,也可以比较从两个多倍体入选物中获得的 SNPs,以寻找 SNP 分歧。
SNiPloid: A Utility to Exploit High-Throughput SNP Data Derived from RNA-Seq in Allopolyploid Species.
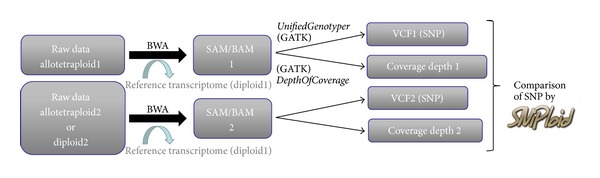
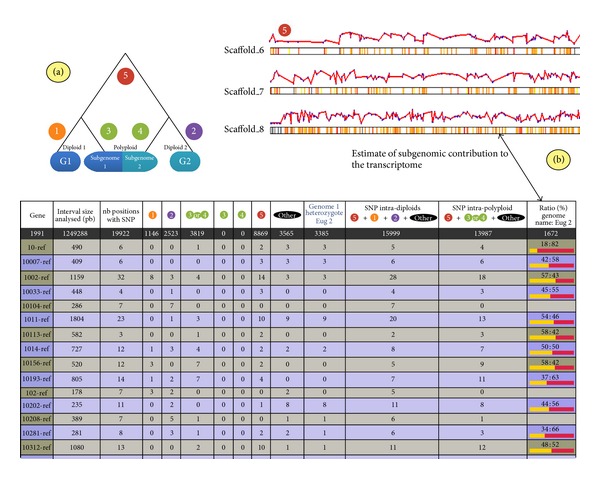
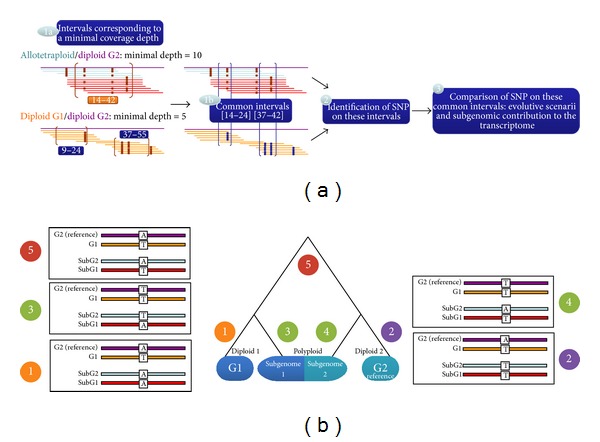
High-throughput sequencing is a common approach to discover SNP variants, especially in plant species. However, methods to analyze predicted SNPs are often optimized for diploid plant species whereas many crop species are allopolyploids and combine related but divergent subgenomes (homoeologous chromosome sets). We created a software tool, SNiPloid, that exploits and interprets putative SNPs in the context of allopolyploidy by comparing SNPs from an allopolyploid with those obtained in its modern-day diploid progenitors. SNiPloid can compare SNPs obtained from a sample to estimate the subgenome contribution to the transcriptome or SNPs obtained from two polyploid accessions to search for SNP divergence.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
