Shopnil Akash, Guendouzi Abdelkrim, Imren Bayil, Md. Eram Hosen, Nobendu Mukerjee, Abdullah F. Shater, Fayez M. Saleh, Ghadeer M. Albadrani, Muath Q. Al-Ghadi, Mohamed M. Abdel-Daim, Tuğba Taşkin Tok
{"title":"通过单倍平修饰发现抗疟疾药物:一种先进的计算方法。","authors":"Shopnil Akash, Guendouzi Abdelkrim, Imren Bayil, Md. Eram Hosen, Nobendu Mukerjee, Abdullah F. Shater, Fayez M. Saleh, Ghadeer M. Albadrani, Muath Q. Al-Ghadi, Mohamed M. Abdel-Daim, Tuğba Taşkin Tok","doi":"10.1111/jcmm.17940","DOIUrl":null,"url":null,"abstract":"<p>The widespread emergence of antimalarial drug resistance has created a major threat to public health. Malaria is a life-threatening infectious disease caused by <i>Plasmodium</i> spp., which includes Apicoplast DNA polymerase and <i>Plasmodium falciparum</i> cysteine protease falcipain-2. These components play a critical role in their life cycle and metabolic pathway, and are involved in the breakdown of erythrocyte hemoglobin in the host, making them promising targets for anti-malarial drug design. Our current study has been designed to explore the potential inhibitors from haplopine derivatives against these two targets using an in silico approach. A total of nine haplopine derivatives were used to perform molecular docking, and the results revealed that Ligands 03 and 05 showed strong binding affinity compared to the control compound atovaquone. Furthermore, these ligand-protein complexes underwent molecular dynamics simulations, and the results demonstrated that the complexes maintained strong stability in terms of RMSD (root mean square deviation), RMSF (root mean square fluctuation), and Rg (radius of gyration) over a 100 ns simulation period. Additionally, PCA (principal component analysis) analysis and the dynamic cross-correlation matrix showed positive outcomes for the protein-ligand complexes. Moreover, the compounds exhibited no violations of the Lipinski rule, and ADMET (absorption, distribution, metabolism, excretion, and toxicity) predictions yielded positive results without indicating any toxicity. Finally, density functional theory (DFT) and molecular electrostatic potential calculations were conducted, revealing that the mentioned derivatives exhibited better stability and outstanding performance. Overall, this computational approach suggests that these haplopine derivatives could serve as a potential source for developing new, effective antimalarial drugs to combat malaria. However, further in vitro or in vivo studies might be conducted to determine their actual effectiveness.</p>","PeriodicalId":15215,"journal":{"name":"Journal of Cellular and Molecular Medicine","volume":"27 20","pages":"3168-3188"},"PeriodicalIF":4.2000,"publicationDate":"2023-09-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/jcmm.17940","citationCount":"5","resultStr":"{\"title\":\"Antimalarial drug discovery against malaria parasites through haplopine modification: An advanced computational approach\",\"authors\":\"Shopnil Akash, Guendouzi Abdelkrim, Imren Bayil, Md. Eram Hosen, Nobendu Mukerjee, Abdullah F. Shater, Fayez M. Saleh, Ghadeer M. Albadrani, Muath Q. Al-Ghadi, Mohamed M. Abdel-Daim, Tuğba Taşkin Tok\",\"doi\":\"10.1111/jcmm.17940\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The widespread emergence of antimalarial drug resistance has created a major threat to public health. Malaria is a life-threatening infectious disease caused by <i>Plasmodium</i> spp., which includes Apicoplast DNA polymerase and <i>Plasmodium falciparum</i> cysteine protease falcipain-2. These components play a critical role in their life cycle and metabolic pathway, and are involved in the breakdown of erythrocyte hemoglobin in the host, making them promising targets for anti-malarial drug design. Our current study has been designed to explore the potential inhibitors from haplopine derivatives against these two targets using an in silico approach. A total of nine haplopine derivatives were used to perform molecular docking, and the results revealed that Ligands 03 and 05 showed strong binding affinity compared to the control compound atovaquone. Furthermore, these ligand-protein complexes underwent molecular dynamics simulations, and the results demonstrated that the complexes maintained strong stability in terms of RMSD (root mean square deviation), RMSF (root mean square fluctuation), and Rg (radius of gyration) over a 100 ns simulation period. Additionally, PCA (principal component analysis) analysis and the dynamic cross-correlation matrix showed positive outcomes for the protein-ligand complexes. Moreover, the compounds exhibited no violations of the Lipinski rule, and ADMET (absorption, distribution, metabolism, excretion, and toxicity) predictions yielded positive results without indicating any toxicity. Finally, density functional theory (DFT) and molecular electrostatic potential calculations were conducted, revealing that the mentioned derivatives exhibited better stability and outstanding performance. Overall, this computational approach suggests that these haplopine derivatives could serve as a potential source for developing new, effective antimalarial drugs to combat malaria. However, further in vitro or in vivo studies might be conducted to determine their actual effectiveness.</p>\",\"PeriodicalId\":15215,\"journal\":{\"name\":\"Journal of Cellular and Molecular Medicine\",\"volume\":\"27 20\",\"pages\":\"3168-3188\"},\"PeriodicalIF\":4.2000,\"publicationDate\":\"2023-09-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/jcmm.17940\",\"citationCount\":\"5\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Cellular and Molecular Medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/jcmm.17940\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Cellular and Molecular Medicine","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/jcmm.17940","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Antimalarial drug discovery against malaria parasites through haplopine modification: An advanced computational approach
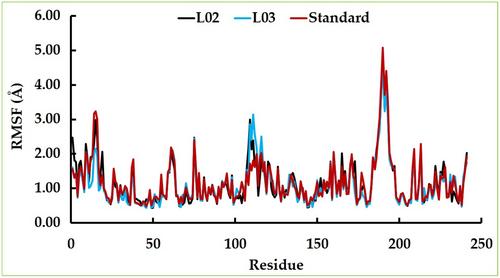
The widespread emergence of antimalarial drug resistance has created a major threat to public health. Malaria is a life-threatening infectious disease caused by Plasmodium spp., which includes Apicoplast DNA polymerase and Plasmodium falciparum cysteine protease falcipain-2. These components play a critical role in their life cycle and metabolic pathway, and are involved in the breakdown of erythrocyte hemoglobin in the host, making them promising targets for anti-malarial drug design. Our current study has been designed to explore the potential inhibitors from haplopine derivatives against these two targets using an in silico approach. A total of nine haplopine derivatives were used to perform molecular docking, and the results revealed that Ligands 03 and 05 showed strong binding affinity compared to the control compound atovaquone. Furthermore, these ligand-protein complexes underwent molecular dynamics simulations, and the results demonstrated that the complexes maintained strong stability in terms of RMSD (root mean square deviation), RMSF (root mean square fluctuation), and Rg (radius of gyration) over a 100 ns simulation period. Additionally, PCA (principal component analysis) analysis and the dynamic cross-correlation matrix showed positive outcomes for the protein-ligand complexes. Moreover, the compounds exhibited no violations of the Lipinski rule, and ADMET (absorption, distribution, metabolism, excretion, and toxicity) predictions yielded positive results without indicating any toxicity. Finally, density functional theory (DFT) and molecular electrostatic potential calculations were conducted, revealing that the mentioned derivatives exhibited better stability and outstanding performance. Overall, this computational approach suggests that these haplopine derivatives could serve as a potential source for developing new, effective antimalarial drugs to combat malaria. However, further in vitro or in vivo studies might be conducted to determine their actual effectiveness.
期刊介绍:
Bridging physiology and cellular medicine, and molecular biology and molecular therapeutics, Journal of Cellular and Molecular Medicine publishes basic research that furthers our understanding of the cellular and molecular mechanisms of disease and translational studies that convert this knowledge into therapeutic approaches.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
