{"title":"埃及患者中严重急性呼吸系统综合征冠状病毒2型的比较基因分型:接近全长的基因组序列与选定的刺突和核衣壳区域。","authors":"Rasha Emad, Iman S Naga","doi":"10.1007/s00430-023-00783-8","DOIUrl":null,"url":null,"abstract":"<p><p>Several tools have been developed for severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) genotyping based on either whole genome or spike sequencing. We aimed to highlight the molecular epidemiological landscape of SARS-CoV-2 in Egypt since the start of the pandemic, to describe discrepancies between the 3 typing tools: Global Initiative on Sharing Avian Influenza Data (GISAID), Nextclade, and Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) and to assess the fitness of spike and nucleocapsid regions for lineage assignment compared to the whole genome. A total of 3935 sequences isolated from Egypt (March 2020-2023) were retrieved from the GISAID database. A subset of data (n = 1212) with high coverage whole genome was used for tool discrimination and agreement analyses. Among 1212 sequences, the highest discriminatory power was 0.895 for PANGOLIN, followed by GISAID (0.872) and Nextclade (0.866). There was a statistically significant difference (p = 0.0418) between lineages assigned via spike (30%) and nucleocapsid (46%) compared to their whole genome-assigned lineages. The first 3 pandemic waves were dominated by B.1, followed by C.36 and then C.36.3, while the fourth to sixth waves were dominated by the B.1.617.2, BA, and BA.5.2 lineages, respectively. Current shift in lineage typing to recombinant forms. The 3 typing tools showed comparable discrimination among SARS-CoV-2 lineages. The nucleocapsid region could be used for lineage assignment.</p>","PeriodicalId":18369,"journal":{"name":"Medical Microbiology and Immunology","volume":" ","pages":"437-446"},"PeriodicalIF":3.0000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10618331/pdf/","citationCount":"0","resultStr":"{\"title\":\"Comparative genotyping of SARS-CoV-2 among Egyptian patients: near-full length genomic sequences versus selected spike and nucleocapsid regions.\",\"authors\":\"Rasha Emad, Iman S Naga\",\"doi\":\"10.1007/s00430-023-00783-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Several tools have been developed for severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) genotyping based on either whole genome or spike sequencing. We aimed to highlight the molecular epidemiological landscape of SARS-CoV-2 in Egypt since the start of the pandemic, to describe discrepancies between the 3 typing tools: Global Initiative on Sharing Avian Influenza Data (GISAID), Nextclade, and Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) and to assess the fitness of spike and nucleocapsid regions for lineage assignment compared to the whole genome. A total of 3935 sequences isolated from Egypt (March 2020-2023) were retrieved from the GISAID database. A subset of data (n = 1212) with high coverage whole genome was used for tool discrimination and agreement analyses. Among 1212 sequences, the highest discriminatory power was 0.895 for PANGOLIN, followed by GISAID (0.872) and Nextclade (0.866). There was a statistically significant difference (p = 0.0418) between lineages assigned via spike (30%) and nucleocapsid (46%) compared to their whole genome-assigned lineages. The first 3 pandemic waves were dominated by B.1, followed by C.36 and then C.36.3, while the fourth to sixth waves were dominated by the B.1.617.2, BA, and BA.5.2 lineages, respectively. Current shift in lineage typing to recombinant forms. The 3 typing tools showed comparable discrimination among SARS-CoV-2 lineages. The nucleocapsid region could be used for lineage assignment.</p>\",\"PeriodicalId\":18369,\"journal\":{\"name\":\"Medical Microbiology and Immunology\",\"volume\":\" \",\"pages\":\"437-446\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2023-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10618331/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Medical Microbiology and Immunology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s00430-023-00783-8\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/10/4 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Medical Microbiology and Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s00430-023-00783-8","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/10/4 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
Comparative genotyping of SARS-CoV-2 among Egyptian patients: near-full length genomic sequences versus selected spike and nucleocapsid regions.
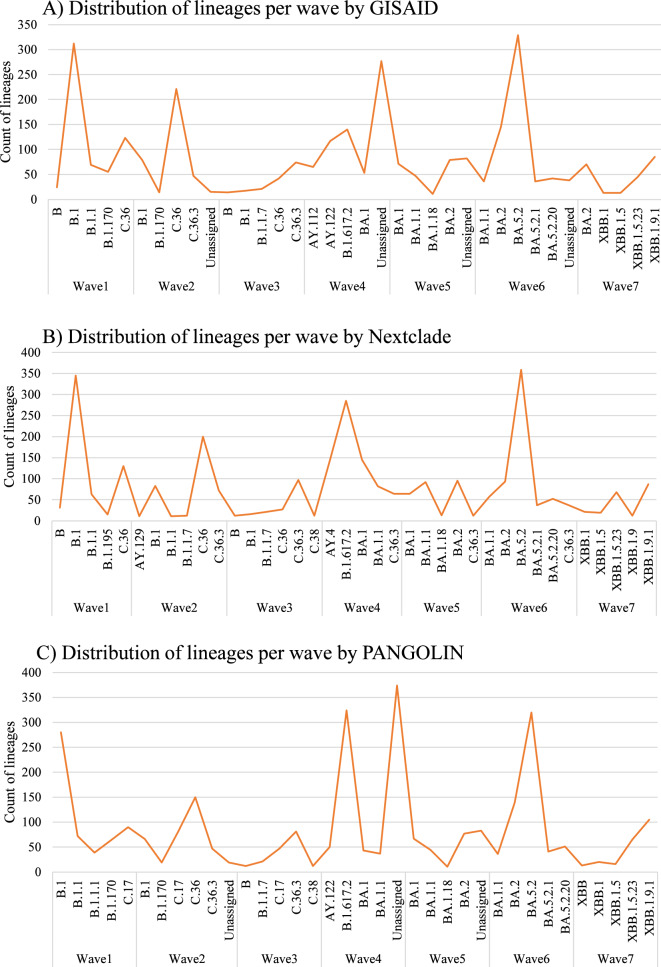
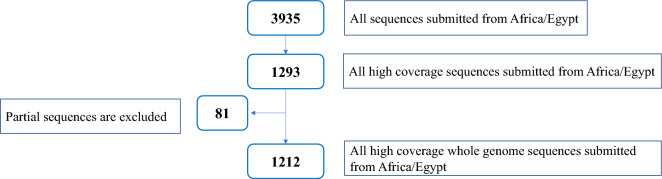
Several tools have been developed for severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) genotyping based on either whole genome or spike sequencing. We aimed to highlight the molecular epidemiological landscape of SARS-CoV-2 in Egypt since the start of the pandemic, to describe discrepancies between the 3 typing tools: Global Initiative on Sharing Avian Influenza Data (GISAID), Nextclade, and Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) and to assess the fitness of spike and nucleocapsid regions for lineage assignment compared to the whole genome. A total of 3935 sequences isolated from Egypt (March 2020-2023) were retrieved from the GISAID database. A subset of data (n = 1212) with high coverage whole genome was used for tool discrimination and agreement analyses. Among 1212 sequences, the highest discriminatory power was 0.895 for PANGOLIN, followed by GISAID (0.872) and Nextclade (0.866). There was a statistically significant difference (p = 0.0418) between lineages assigned via spike (30%) and nucleocapsid (46%) compared to their whole genome-assigned lineages. The first 3 pandemic waves were dominated by B.1, followed by C.36 and then C.36.3, while the fourth to sixth waves were dominated by the B.1.617.2, BA, and BA.5.2 lineages, respectively. Current shift in lineage typing to recombinant forms. The 3 typing tools showed comparable discrimination among SARS-CoV-2 lineages. The nucleocapsid region could be used for lineage assignment.
期刊介绍:
Medical Microbiology and Immunology (MMIM) publishes key findings on all aspects of the interrelationship between infectious agents and the immune system of their hosts. The journal´s main focus is original research work on intrinsic, innate or adaptive immune responses to viral, bacterial, fungal and parasitic (protozoan and helminthic) infections and on the virulence of the respective infectious pathogens.
MMIM covers basic, translational as well as clinical research in infectious diseases and infectious disease immunology. Basic research using cell cultures, organoid, and animal models are welcome, provided that the models have a clinical correlate and address a relevant medical question.
The journal also considers manuscripts on the epidemiology of infectious diseases, including the emergence and epidemic spreading of pathogens and the development of resistance to anti-infective therapies, and on novel vaccines and other innovative measurements of prevention.
The following categories of manuscripts will not be considered for publication in MMIM:
submissions of preliminary work, of merely descriptive data sets without investigation of mechanisms or of limited global interest,
manuscripts on existing or novel anti-infective compounds, which focus on pharmaceutical or pharmacological aspects of the drugs,
manuscripts on existing or modified vaccines, unless they report on experimental or clinical efficacy studies or provide new immunological information on their mode of action,
manuscripts on the diagnostics of infectious diseases, unless they offer a novel concept to solve a pending diagnostic problem,
case reports or case series, unless they are embedded in a study that focuses on the anti-infectious immune response and/or on the virulence of a pathogen.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
