{"title":"药物发现中结合自由能计算的缓慢而稳定的增长","authors":"Huafeng Xu","doi":"10.1007/s10822-022-00494-x","DOIUrl":null,"url":null,"abstract":"<div><p>Binding free energy calculations are increasingly used in drug discovery research to predict protein-ligand binding affinities and to prioritize candidate drug molecules accordingly. It has taken decades of collective effort to transform this academic concept into a technology adopted by the pharmaceutical and biotech industry. Having personally witnessed and taken part in this transformation, here I recount the (incomplete) list of problems that had to be solved to make this computational tool practical and suggest areas of future development.</p></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"37 2","pages":"67 - 74"},"PeriodicalIF":3.1000,"publicationDate":"2022-12-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"6","resultStr":"{\"title\":\"The slow but steady rise of binding free energy calculations in drug discovery\",\"authors\":\"Huafeng Xu\",\"doi\":\"10.1007/s10822-022-00494-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Binding free energy calculations are increasingly used in drug discovery research to predict protein-ligand binding affinities and to prioritize candidate drug molecules accordingly. It has taken decades of collective effort to transform this academic concept into a technology adopted by the pharmaceutical and biotech industry. Having personally witnessed and taken part in this transformation, here I recount the (incomplete) list of problems that had to be solved to make this computational tool practical and suggest areas of future development.</p></div>\",\"PeriodicalId\":621,\"journal\":{\"name\":\"Journal of Computer-Aided Molecular Design\",\"volume\":\"37 2\",\"pages\":\"67 - 74\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2022-12-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"6\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computer-Aided Molecular Design\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10822-022-00494-x\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-022-00494-x","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
The slow but steady rise of binding free energy calculations in drug discovery
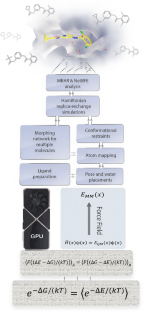
Binding free energy calculations are increasingly used in drug discovery research to predict protein-ligand binding affinities and to prioritize candidate drug molecules accordingly. It has taken decades of collective effort to transform this academic concept into a technology adopted by the pharmaceutical and biotech industry. Having personally witnessed and taken part in this transformation, here I recount the (incomplete) list of problems that had to be solved to make this computational tool practical and suggest areas of future development.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
