Deniz Ece Kaya, Ege Ülgen, Ayşe Sesin Kocagöz, Osman Uğur Sezerman
{"title":"各种特征提取和机器学习方法在肺炎链球菌耐药预测中的比较","authors":"Deniz Ece Kaya, Ege Ülgen, Ayşe Sesin Kocagöz, Osman Uğur Sezerman","doi":"10.3389/frabi.2023.1126468","DOIUrl":null,"url":null,"abstract":"<p><p>Streptococcus pneumoniae is one of the major concerns of clinicians and one of the global public health problems. This pathogen is associated with high morbidity and mortality rates and antimicrobial resistance (AMR). In the last few years, reduced genome sequencing costs have made it possible to explore more of the drug resistance of S. pneumoniae, and machine learning (ML) has become a popular tool for understanding, diagnosing, treating, and predicting these phenotypes. Nucleotide k-mers, amino acid k-mers, single nucleotide polymorphisms (SNPs), and combinations of these features have rich genetic information in whole-genome sequencing. This study compares different ML models for predicting AMR phenotype for S. pneumoniae. We compared nucleotide k-mers, amino acid k-mers, SNPs, and their combinations to predict AMR in S. pneumoniae for three antibiotics: Penicillin, Erythromycin, and Tetracycline. 980 pneumococcal strains were downloaded from the European Nucleotide Archive (ENA). Furthermore, we used and compared several machine learning methods to train the models, including random forests, support vector machines, stochastic gradient boosting, and extreme gradient boosting. In this study, we found that key features of the AMR prediction model setup and the choice of machine learning method affected the results. The approach can be applied here to further studies to improve AMR prediction accuracy and efficiency.</p>","PeriodicalId":73065,"journal":{"name":"Frontiers in antibiotics","volume":" ","pages":"1126468"},"PeriodicalIF":0.0000,"publicationDate":"2023-03-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11731958/pdf/","citationCount":"0","resultStr":"{\"title\":\"A comparison of various feature extraction and machine learning methods for antimicrobial resistance prediction in <i>streptococcus pneumoniae</i>.\",\"authors\":\"Deniz Ece Kaya, Ege Ülgen, Ayşe Sesin Kocagöz, Osman Uğur Sezerman\",\"doi\":\"10.3389/frabi.2023.1126468\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Streptococcus pneumoniae is one of the major concerns of clinicians and one of the global public health problems. This pathogen is associated with high morbidity and mortality rates and antimicrobial resistance (AMR). In the last few years, reduced genome sequencing costs have made it possible to explore more of the drug resistance of S. pneumoniae, and machine learning (ML) has become a popular tool for understanding, diagnosing, treating, and predicting these phenotypes. Nucleotide k-mers, amino acid k-mers, single nucleotide polymorphisms (SNPs), and combinations of these features have rich genetic information in whole-genome sequencing. This study compares different ML models for predicting AMR phenotype for S. pneumoniae. We compared nucleotide k-mers, amino acid k-mers, SNPs, and their combinations to predict AMR in S. pneumoniae for three antibiotics: Penicillin, Erythromycin, and Tetracycline. 980 pneumococcal strains were downloaded from the European Nucleotide Archive (ENA). Furthermore, we used and compared several machine learning methods to train the models, including random forests, support vector machines, stochastic gradient boosting, and extreme gradient boosting. In this study, we found that key features of the AMR prediction model setup and the choice of machine learning method affected the results. The approach can be applied here to further studies to improve AMR prediction accuracy and efficiency.</p>\",\"PeriodicalId\":73065,\"journal\":{\"name\":\"Frontiers in antibiotics\",\"volume\":\" \",\"pages\":\"1126468\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-03-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11731958/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Frontiers in antibiotics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3389/frabi.2023.1126468\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in antibiotics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/frabi.2023.1126468","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
A comparison of various feature extraction and machine learning methods for antimicrobial resistance prediction in streptococcus pneumoniae.
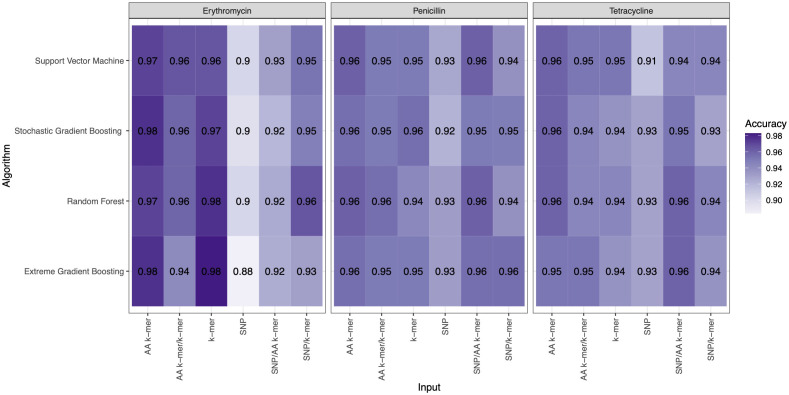
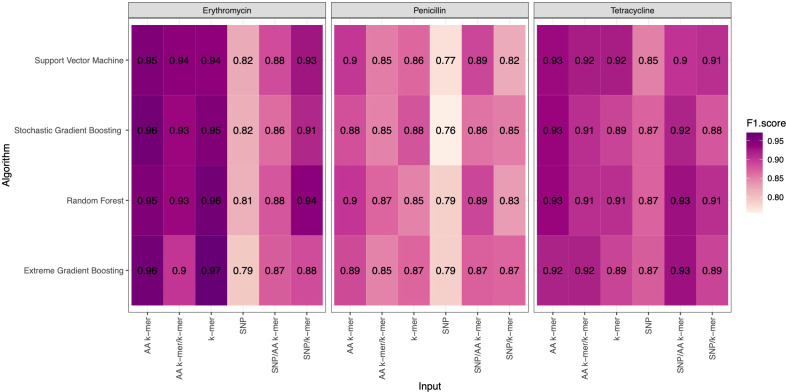
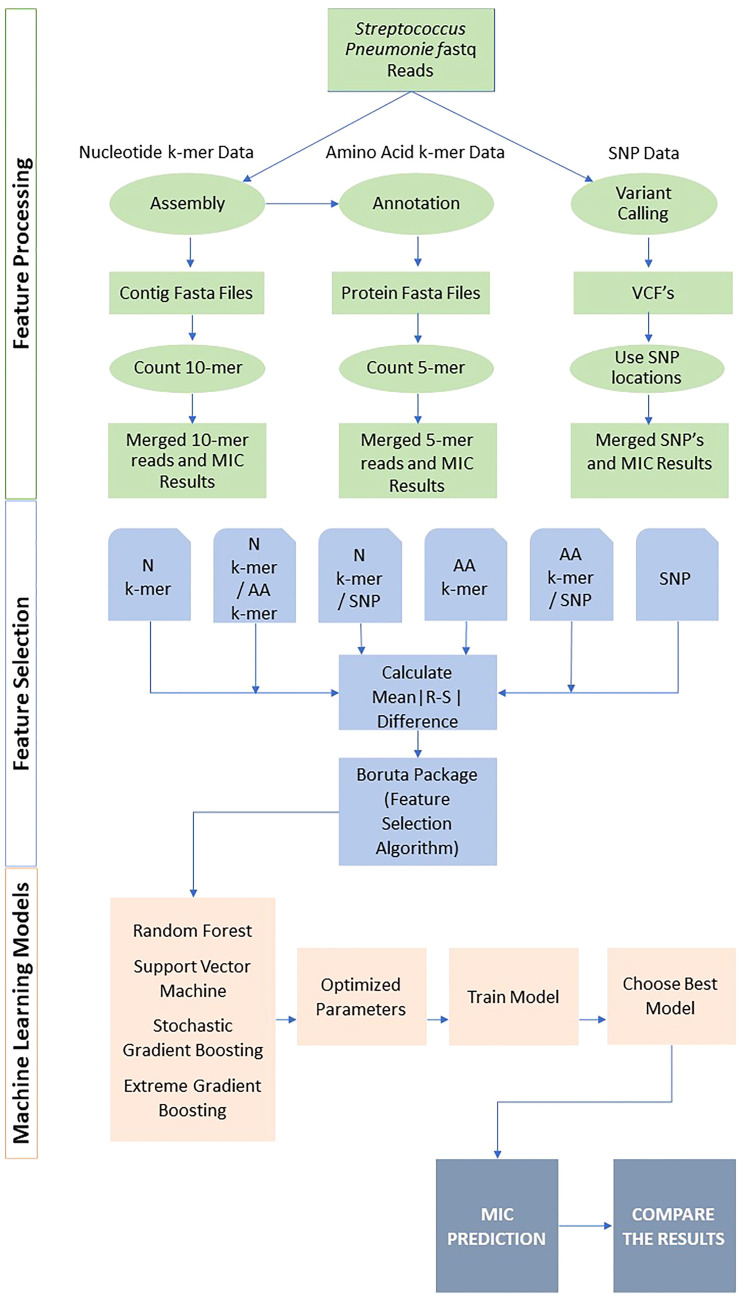
Streptococcus pneumoniae is one of the major concerns of clinicians and one of the global public health problems. This pathogen is associated with high morbidity and mortality rates and antimicrobial resistance (AMR). In the last few years, reduced genome sequencing costs have made it possible to explore more of the drug resistance of S. pneumoniae, and machine learning (ML) has become a popular tool for understanding, diagnosing, treating, and predicting these phenotypes. Nucleotide k-mers, amino acid k-mers, single nucleotide polymorphisms (SNPs), and combinations of these features have rich genetic information in whole-genome sequencing. This study compares different ML models for predicting AMR phenotype for S. pneumoniae. We compared nucleotide k-mers, amino acid k-mers, SNPs, and their combinations to predict AMR in S. pneumoniae for three antibiotics: Penicillin, Erythromycin, and Tetracycline. 980 pneumococcal strains were downloaded from the European Nucleotide Archive (ENA). Furthermore, we used and compared several machine learning methods to train the models, including random forests, support vector machines, stochastic gradient boosting, and extreme gradient boosting. In this study, we found that key features of the AMR prediction model setup and the choice of machine learning method affected the results. The approach can be applied here to further studies to improve AMR prediction accuracy and efficiency.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
