Nayanthara K. Jayadev, Sudipta Roy, Ashwani K Tiwari
{"title":"Pd(111)表面NO + CO反应的计算研究:晶格位置对吸附和反应性的影响","authors":"Nayanthara K. Jayadev, Sudipta Roy, Ashwani K Tiwari","doi":"10.1007/s12039-023-02178-x","DOIUrl":null,"url":null,"abstract":"<div><p>The NO + CO reaction is considered a prototype reaction that portrays the abatement of NOx, the primary pollutant from automobile exhausts, catalyzed by metals like Pd, Pt, and Rh. The detailed mechanism of this reaction on the Pd(111) surface was devised by taking into account all probable elementary reactions with the aid of density functional theory calculations. The reaction steps under consideration include NO dissociation, N<sub>2</sub> formation from N-N recombination, CO<sub>2</sub> formation, N<sub>2</sub>O formation, and N<sub>2</sub>O dissociation, respectively. The adsorption energies, reaction energies, and activation energy barriers were calculated for all the elementary steps of the reaction. Depending on the lattice site, over which the intermediates of the elementary steps get adsorbed, adsorption energies vary significantly. Both N-N recombination and N<sub>2</sub>O decomposition were identified as the reaction pathways for N<sub>2</sub> formation. The NO dissociation step could be regarded as the rate-determining step, bearing the highest activation energy barrier among all the elementary reactions. The reactivity of this step has been shown to increase with an increase in the surface temperature. It was also observed that N<sub>2</sub> formation from N-N recombination controls the overall reaction rate to a certain extent. The influence of different Pd facets on NO dissociation was scrutinized, and it was found that (100) facet exhibits a lower activation energy barrier than (111) metal surface. The higher activity of (100) surface was explained with the help of the density of states plots.</p><h3>Graphical abstract</h3><p>The model reaction NO + CO has been studied in detail on Pd (111) surface to understand Three Way Catalytic (TWC) converters. NO dissociation step was found to be the RDS. The surface temperature has been shown to affect the reactivity of the RDS. Pd(100) could be used for better performance.\n</p><figure><div><div><div><picture><source><img></source></picture></div></div></div></figure></div>","PeriodicalId":50242,"journal":{"name":"Journal of Chemical Sciences","volume":"135 3","pages":""},"PeriodicalIF":2.0000,"publicationDate":"2023-06-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Computational study of NO + CO reaction on Pd(111) surface: effect of lattice sites on the adsorption and reactivity\",\"authors\":\"Nayanthara K. Jayadev, Sudipta Roy, Ashwani K Tiwari\",\"doi\":\"10.1007/s12039-023-02178-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The NO + CO reaction is considered a prototype reaction that portrays the abatement of NOx, the primary pollutant from automobile exhausts, catalyzed by metals like Pd, Pt, and Rh. The detailed mechanism of this reaction on the Pd(111) surface was devised by taking into account all probable elementary reactions with the aid of density functional theory calculations. The reaction steps under consideration include NO dissociation, N<sub>2</sub> formation from N-N recombination, CO<sub>2</sub> formation, N<sub>2</sub>O formation, and N<sub>2</sub>O dissociation, respectively. The adsorption energies, reaction energies, and activation energy barriers were calculated for all the elementary steps of the reaction. Depending on the lattice site, over which the intermediates of the elementary steps get adsorbed, adsorption energies vary significantly. Both N-N recombination and N<sub>2</sub>O decomposition were identified as the reaction pathways for N<sub>2</sub> formation. The NO dissociation step could be regarded as the rate-determining step, bearing the highest activation energy barrier among all the elementary reactions. The reactivity of this step has been shown to increase with an increase in the surface temperature. It was also observed that N<sub>2</sub> formation from N-N recombination controls the overall reaction rate to a certain extent. The influence of different Pd facets on NO dissociation was scrutinized, and it was found that (100) facet exhibits a lower activation energy barrier than (111) metal surface. The higher activity of (100) surface was explained with the help of the density of states plots.</p><h3>Graphical abstract</h3><p>The model reaction NO + CO has been studied in detail on Pd (111) surface to understand Three Way Catalytic (TWC) converters. NO dissociation step was found to be the RDS. The surface temperature has been shown to affect the reactivity of the RDS. Pd(100) could be used for better performance.\\n</p><figure><div><div><div><picture><source><img></source></picture></div></div></div></figure></div>\",\"PeriodicalId\":50242,\"journal\":{\"name\":\"Journal of Chemical Sciences\",\"volume\":\"135 3\",\"pages\":\"\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2023-06-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Sciences\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s12039-023-02178-x\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"Chemistry\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Sciences","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s12039-023-02178-x","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Chemistry","Score":null,"Total":0}
引用次数: 0
摘要
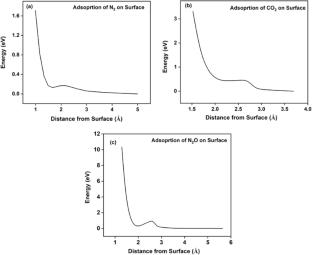
NO + CO反应被认为是一个原型反应,描绘了减少氮氧化物(来自汽车尾气的主要污染物),由钯、铂和铑等金属催化。在密度泛函理论计算的帮助下,考虑了所有可能的基本反应,设计了Pd(111)表面上该反应的详细机理。考虑的反应步骤分别为NO解离、N-N重组生成N2、CO2生成、N2O生成和N2O解离。计算了反应各基本步骤的吸附能、反应能和活化能势垒。根据基本步骤中间产物被吸附的晶格位置的不同,吸附能变化很大。N-N重组和N2O分解都是N2生成的反应途径。在所有的元素反应中,NO的解离反应具有最高的活化能势垒,是反应的速率决定步骤。这一步骤的反应活性随着表面温度的升高而增加。N-N重组生成N2在一定程度上控制了总反应速率。研究了不同Pd面对NO解离的影响,发现(100)面比(111)金属表面具有更低的活化能垒。用态密度图解释了(100)表面活性较高的原因。为了更好地理解三元催化(TWC)转化器,对Pd(111)表面NO + CO的模型反应进行了详细的研究。没有发现解离步骤是RDS。表面温度对RDS的反应性有影响。使用Pd(100)可以获得更好的性能。
Computational study of NO + CO reaction on Pd(111) surface: effect of lattice sites on the adsorption and reactivity
The NO + CO reaction is considered a prototype reaction that portrays the abatement of NOx, the primary pollutant from automobile exhausts, catalyzed by metals like Pd, Pt, and Rh. The detailed mechanism of this reaction on the Pd(111) surface was devised by taking into account all probable elementary reactions with the aid of density functional theory calculations. The reaction steps under consideration include NO dissociation, N2 formation from N-N recombination, CO2 formation, N2O formation, and N2O dissociation, respectively. The adsorption energies, reaction energies, and activation energy barriers were calculated for all the elementary steps of the reaction. Depending on the lattice site, over which the intermediates of the elementary steps get adsorbed, adsorption energies vary significantly. Both N-N recombination and N2O decomposition were identified as the reaction pathways for N2 formation. The NO dissociation step could be regarded as the rate-determining step, bearing the highest activation energy barrier among all the elementary reactions. The reactivity of this step has been shown to increase with an increase in the surface temperature. It was also observed that N2 formation from N-N recombination controls the overall reaction rate to a certain extent. The influence of different Pd facets on NO dissociation was scrutinized, and it was found that (100) facet exhibits a lower activation energy barrier than (111) metal surface. The higher activity of (100) surface was explained with the help of the density of states plots.
Graphical abstract
The model reaction NO + CO has been studied in detail on Pd (111) surface to understand Three Way Catalytic (TWC) converters. NO dissociation step was found to be the RDS. The surface temperature has been shown to affect the reactivity of the RDS. Pd(100) could be used for better performance.
期刊介绍:
Journal of Chemical Sciences is a monthly journal published by the Indian Academy of Sciences. It formed part of the original Proceedings of the Indian Academy of Sciences – Part A, started by the Nobel Laureate Prof C V Raman in 1934, that was split in 1978 into three separate journals. It was renamed as Journal of Chemical Sciences in 2004. The journal publishes original research articles and rapid communications, covering all areas of chemical sciences. A significant feature of the journal is its special issues, brought out from time to time, devoted to conference symposia/proceedings in frontier areas of the subject, held not only in India but also in other countries.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
