Jinhong Ren, Tasneem M. Vaid, Hyun Lee, Isabel Ojeda, Michael E. Johnson
{"title":"利用分子对接、分子动力学模拟和结合自由能计算评估丙型肝炎病毒NS3/4A与磺胺基苯甲酰胺分子之间的相互作用","authors":"Jinhong Ren, Tasneem M. Vaid, Hyun Lee, Isabel Ojeda, Michael E. Johnson","doi":"10.1007/s10822-022-00490-1","DOIUrl":null,"url":null,"abstract":"<div><p>The Hepatitis C Virus (HCV) NS3/4A is an attractive target for the treatment of Hepatitis C infection. Herein, we present an investigation of HCV NS3/4A inhibitors based on a sulfonamidobenzamide scaffold. Inhibitor interactions with HCV NS3/4A were explored by molecular docking, molecular dynamics simulations, and MM/PBSA binding free energy calculations. All of the inhibitors adopt similar molecular docking poses in the catalytic site of the protease that are stabilized by hydrogen bond interactions with G137 and the catalytic S139, which are known to be important for potency and binding stability. The quantitative assessments of binding free energies from MM/PBSA correlate well with the experimental results, with a high coefficient of determination, R<sup>2</sup> of 0.92. Binding free energy decomposition analyses elucidate the different contributions of Q41, F43, H57, R109, K136, G137, S138, S139, A156, M485, and Q526 in binding different inhibitors. The importance of these sidechain contributions was further confirmed by computational alanine scanning mutagenesis. In addition, the sidechains of K136 and S139 show crucial but distinct contributions to inhibitor binding with HCV NS3/4A. The structural basis of the potency has been elucidated, demonstrating the importance of the R155 sidechain conformation. This extensive exploration of binding energies and interactions between these compounds and HCV NS3/4A at the atomic level should benefit future antiviral drug design.</p></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"37 1","pages":"53 - 65"},"PeriodicalIF":3.1000,"publicationDate":"2022-11-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s10822-022-00490-1.pdf","citationCount":"2","resultStr":"{\"title\":\"Evaluation of interactions between the hepatitis C virus NS3/4A and sulfonamidobenzamide based molecules using molecular docking, molecular dynamics simulations and binding free energy calculations\",\"authors\":\"Jinhong Ren, Tasneem M. Vaid, Hyun Lee, Isabel Ojeda, Michael E. Johnson\",\"doi\":\"10.1007/s10822-022-00490-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The Hepatitis C Virus (HCV) NS3/4A is an attractive target for the treatment of Hepatitis C infection. Herein, we present an investigation of HCV NS3/4A inhibitors based on a sulfonamidobenzamide scaffold. Inhibitor interactions with HCV NS3/4A were explored by molecular docking, molecular dynamics simulations, and MM/PBSA binding free energy calculations. All of the inhibitors adopt similar molecular docking poses in the catalytic site of the protease that are stabilized by hydrogen bond interactions with G137 and the catalytic S139, which are known to be important for potency and binding stability. The quantitative assessments of binding free energies from MM/PBSA correlate well with the experimental results, with a high coefficient of determination, R<sup>2</sup> of 0.92. Binding free energy decomposition analyses elucidate the different contributions of Q41, F43, H57, R109, K136, G137, S138, S139, A156, M485, and Q526 in binding different inhibitors. The importance of these sidechain contributions was further confirmed by computational alanine scanning mutagenesis. In addition, the sidechains of K136 and S139 show crucial but distinct contributions to inhibitor binding with HCV NS3/4A. The structural basis of the potency has been elucidated, demonstrating the importance of the R155 sidechain conformation. This extensive exploration of binding energies and interactions between these compounds and HCV NS3/4A at the atomic level should benefit future antiviral drug design.</p></div>\",\"PeriodicalId\":621,\"journal\":{\"name\":\"Journal of Computer-Aided Molecular Design\",\"volume\":\"37 1\",\"pages\":\"53 - 65\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2022-11-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://link.springer.com/content/pdf/10.1007/s10822-022-00490-1.pdf\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computer-Aided Molecular Design\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10822-022-00490-1\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-022-00490-1","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Evaluation of interactions between the hepatitis C virus NS3/4A and sulfonamidobenzamide based molecules using molecular docking, molecular dynamics simulations and binding free energy calculations
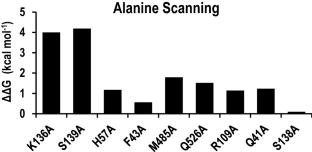
The Hepatitis C Virus (HCV) NS3/4A is an attractive target for the treatment of Hepatitis C infection. Herein, we present an investigation of HCV NS3/4A inhibitors based on a sulfonamidobenzamide scaffold. Inhibitor interactions with HCV NS3/4A were explored by molecular docking, molecular dynamics simulations, and MM/PBSA binding free energy calculations. All of the inhibitors adopt similar molecular docking poses in the catalytic site of the protease that are stabilized by hydrogen bond interactions with G137 and the catalytic S139, which are known to be important for potency and binding stability. The quantitative assessments of binding free energies from MM/PBSA correlate well with the experimental results, with a high coefficient of determination, R2 of 0.92. Binding free energy decomposition analyses elucidate the different contributions of Q41, F43, H57, R109, K136, G137, S138, S139, A156, M485, and Q526 in binding different inhibitors. The importance of these sidechain contributions was further confirmed by computational alanine scanning mutagenesis. In addition, the sidechains of K136 and S139 show crucial but distinct contributions to inhibitor binding with HCV NS3/4A. The structural basis of the potency has been elucidated, demonstrating the importance of the R155 sidechain conformation. This extensive exploration of binding energies and interactions between these compounds and HCV NS3/4A at the atomic level should benefit future antiviral drug design.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
