Julieta Reyna-Luna, Luis Soriano-Agueda, Christiaan Jardinez Vera, Marco Franco-Pérez
{"title":"抗肿瘤药物多柔比星与 3d 过渡金属离子 Zn2+、Cu2+ 和 VO2+ 的配位化学透视:使用校准良好的热力学循环和化学相互作用量子化学模型进行的研究","authors":"Julieta Reyna-Luna, Luis Soriano-Agueda, Christiaan Jardinez Vera, Marco Franco-Pérez","doi":"10.1007/s10822-023-00506-4","DOIUrl":null,"url":null,"abstract":"<div><p>We present a computational strategy based on thermodynamic cycles to predict and describe the chemical equilibrium between the 3<i>d</i>-transition metal ions Zn<sup>2+</sup>, Cu<sup>2+</sup>, and VO<sup>2+</sup> and the widely used antineoplastic drug doxorubicin. Our method involves benchmarking a theoretical protocol to compute gas-phase quantities using DLPNO Coupled-Cluster calculations as reference, followed by estimating solvation contributions to the reaction Gibbs free energies using both explicit partial (micro)solvation steps for charged solutes and neutral coordination complexes, as well as a continuum solvation procedure for all solutes involved in the complexation process. We rationalized the stability of these doxorubicin-metal complexes by inspecting quantities obtained from the topology of their electron densities, particularly the bond critical points and non-covalent interaction index. Our approach allowed us to identify representative species in solution phase, infer the most likely complexation process for each case, and identify key intramolecular interactions involved in the stability of these compounds. To the best of our knowledge, this is the first study reporting thermodynamic constants for the complexation of doxorubicin with transition metal ions. Unlike other methods, our procedure is computationally affordable for medium-sized systems and provides valuable insights even with limited experimental data. Furthermore, it can be extended to describe the complexation process between 3<i>d-</i>transition metal ions and other bioactive ligands.\n</p></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"37 7","pages":"279 - 299"},"PeriodicalIF":3.1000,"publicationDate":"2023-05-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Insights into the coordination chemistry of antineoplastic doxorubicin with 3d-transition metal ions Zn2+, Cu2+, and VO2+: a study using well-calibrated thermodynamic cycles and chemical interaction quantum chemistry models\",\"authors\":\"Julieta Reyna-Luna, Luis Soriano-Agueda, Christiaan Jardinez Vera, Marco Franco-Pérez\",\"doi\":\"10.1007/s10822-023-00506-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>We present a computational strategy based on thermodynamic cycles to predict and describe the chemical equilibrium between the 3<i>d</i>-transition metal ions Zn<sup>2+</sup>, Cu<sup>2+</sup>, and VO<sup>2+</sup> and the widely used antineoplastic drug doxorubicin. Our method involves benchmarking a theoretical protocol to compute gas-phase quantities using DLPNO Coupled-Cluster calculations as reference, followed by estimating solvation contributions to the reaction Gibbs free energies using both explicit partial (micro)solvation steps for charged solutes and neutral coordination complexes, as well as a continuum solvation procedure for all solutes involved in the complexation process. We rationalized the stability of these doxorubicin-metal complexes by inspecting quantities obtained from the topology of their electron densities, particularly the bond critical points and non-covalent interaction index. Our approach allowed us to identify representative species in solution phase, infer the most likely complexation process for each case, and identify key intramolecular interactions involved in the stability of these compounds. To the best of our knowledge, this is the first study reporting thermodynamic constants for the complexation of doxorubicin with transition metal ions. Unlike other methods, our procedure is computationally affordable for medium-sized systems and provides valuable insights even with limited experimental data. Furthermore, it can be extended to describe the complexation process between 3<i>d-</i>transition metal ions and other bioactive ligands.\\n</p></div>\",\"PeriodicalId\":621,\"journal\":{\"name\":\"Journal of Computer-Aided Molecular Design\",\"volume\":\"37 7\",\"pages\":\"279 - 299\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2023-05-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computer-Aided Molecular Design\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10822-023-00506-4\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-023-00506-4","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
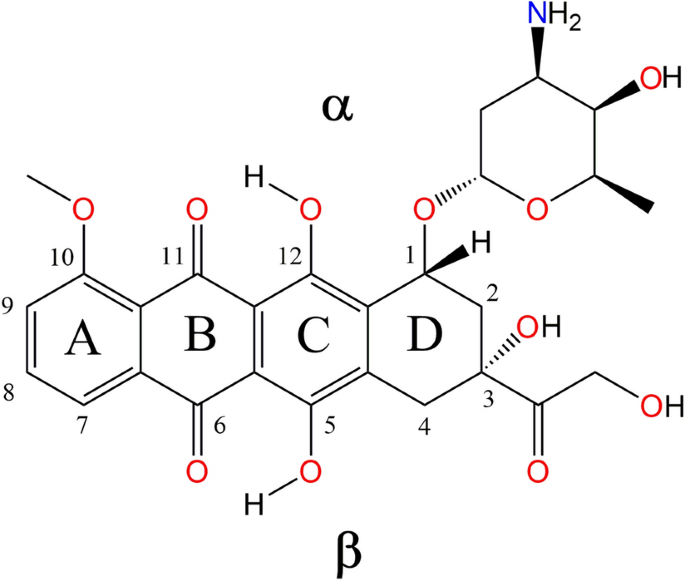
我们提出了一种基于热力学循环的计算策略,用于预测和描述 3d 过渡金属离子 Zn2+、Cu2+ 和 VO2+ 与广泛使用的抗肿瘤药物多柔比星之间的化学平衡。我们的方法包括使用 DLPNO 耦合簇计算作为参考基准来计算气相量,然后使用带电溶质和中性配位复合物的显式部分(微)溶解步骤以及复合物过程中所有溶质的连续溶解程序来估算溶解对反应吉布斯自由能的贡献。我们通过检查从电子密度拓扑结构中获得的量,特别是键临界点和非共价相互作用指数,合理地解释了这些多柔比星-金属复合物的稳定性。通过这种方法,我们可以确定溶液相中的代表性物种,推断出每种情况下最可能的络合过程,并确定涉及这些化合物稳定性的关键分子内相互作用。据我们所知,这是第一项报告多柔比星与过渡金属离子络合热力学常数的研究。与其他方法不同的是,我们的程序在计算上对中等规模的系统是可承受的,即使实验数据有限,也能提供有价值的见解。此外,它还可扩展用于描述 3d 过渡金属离子与其他生物活性配体的复配过程。
Insights into the coordination chemistry of antineoplastic doxorubicin with 3d-transition metal ions Zn2+, Cu2+, and VO2+: a study using well-calibrated thermodynamic cycles and chemical interaction quantum chemistry models
We present a computational strategy based on thermodynamic cycles to predict and describe the chemical equilibrium between the 3d-transition metal ions Zn2+, Cu2+, and VO2+ and the widely used antineoplastic drug doxorubicin. Our method involves benchmarking a theoretical protocol to compute gas-phase quantities using DLPNO Coupled-Cluster calculations as reference, followed by estimating solvation contributions to the reaction Gibbs free energies using both explicit partial (micro)solvation steps for charged solutes and neutral coordination complexes, as well as a continuum solvation procedure for all solutes involved in the complexation process. We rationalized the stability of these doxorubicin-metal complexes by inspecting quantities obtained from the topology of their electron densities, particularly the bond critical points and non-covalent interaction index. Our approach allowed us to identify representative species in solution phase, infer the most likely complexation process for each case, and identify key intramolecular interactions involved in the stability of these compounds. To the best of our knowledge, this is the first study reporting thermodynamic constants for the complexation of doxorubicin with transition metal ions. Unlike other methods, our procedure is computationally affordable for medium-sized systems and provides valuable insights even with limited experimental data. Furthermore, it can be extended to describe the complexation process between 3d-transition metal ions and other bioactive ligands.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
