{"title":"利用原始模型对离子系统进行嵌套蒙特卡罗模拟,将德拜-胡克尔(DH)势能作为重要函数,并利用库尔巴克-莱布勒发散最小化法优化 DH 势能","authors":"Rakesh Srivastava, Pradipta Bandyopadhyay","doi":"10.1007/s12039-023-02167-0","DOIUrl":null,"url":null,"abstract":"<div><p>In this work, Nested Monte Carlo (NMC) simulation was done for symmetric and asymmetric ionic systems where the energy function is described by a combination of hard-sphere and Coulomb interaction (the primitive model). In the NMC method, Monte Carlo (MC) moves for the primary chain (where the energy function is the primitive model) is given by running a short MC trajectory using an auxiliary potential reducing the computational cost significantly. The Debye-Hückel (DH) potential was used as the auxiliary potential in our work. It is shown that with a careful choice of Debye length in the DH potential, and length of the short MC run in the auxiliary chain, the NMC method gives the same result as the more expensive MC simulation using full Ewald summation in the primitive model. Implementing the minimization of the Kullback–Leibler (KL) divergence between the pair-correlation function of the standard (without any auxiliary potential) MC simulation and NMC, a simple algorithm was also presented to develop good DH-like potentials to be used as auxiliary potentials in the nested MC run. Overall, this technique significantly reduces the Ewald summation technique's computational cost and can be applied to any atomic and molecular system with coulombic interaction in the potential energy function.</p><h3>Graphical abstract</h3><p>Nested Monte Carlo (NMC) simulations for symmetric and asymmetric ionic systems are performed, where Monte Carlo (MC) moves for the primary chain are given by running a short MC trajectory using an auxiliary potential. Excellent agreement has been shown between the pair correlation functions calculated from NMC, and standard Ewald simulations.</p><figure><div><div><div><picture><source><img></source></picture></div></div></div></figure></div>","PeriodicalId":50242,"journal":{"name":"Journal of Chemical Sciences","volume":"135 2","pages":""},"PeriodicalIF":2.0000,"publicationDate":"2023-05-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Nested Monte Carlo simulation of ionic systems with the primitive model using Debye-Hückel (DH) potential as an importance function and optimizing the DH potential with Kullback-Leibler divergence minimization\",\"authors\":\"Rakesh Srivastava, Pradipta Bandyopadhyay\",\"doi\":\"10.1007/s12039-023-02167-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>In this work, Nested Monte Carlo (NMC) simulation was done for symmetric and asymmetric ionic systems where the energy function is described by a combination of hard-sphere and Coulomb interaction (the primitive model). In the NMC method, Monte Carlo (MC) moves for the primary chain (where the energy function is the primitive model) is given by running a short MC trajectory using an auxiliary potential reducing the computational cost significantly. The Debye-Hückel (DH) potential was used as the auxiliary potential in our work. It is shown that with a careful choice of Debye length in the DH potential, and length of the short MC run in the auxiliary chain, the NMC method gives the same result as the more expensive MC simulation using full Ewald summation in the primitive model. Implementing the minimization of the Kullback–Leibler (KL) divergence between the pair-correlation function of the standard (without any auxiliary potential) MC simulation and NMC, a simple algorithm was also presented to develop good DH-like potentials to be used as auxiliary potentials in the nested MC run. Overall, this technique significantly reduces the Ewald summation technique's computational cost and can be applied to any atomic and molecular system with coulombic interaction in the potential energy function.</p><h3>Graphical abstract</h3><p>Nested Monte Carlo (NMC) simulations for symmetric and asymmetric ionic systems are performed, where Monte Carlo (MC) moves for the primary chain are given by running a short MC trajectory using an auxiliary potential. Excellent agreement has been shown between the pair correlation functions calculated from NMC, and standard Ewald simulations.</p><figure><div><div><div><picture><source><img></source></picture></div></div></div></figure></div>\",\"PeriodicalId\":50242,\"journal\":{\"name\":\"Journal of Chemical Sciences\",\"volume\":\"135 2\",\"pages\":\"\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2023-05-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Sciences\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s12039-023-02167-0\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"Chemistry\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Sciences","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s12039-023-02167-0","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Chemistry","Score":null,"Total":0}
引用次数: 0
摘要
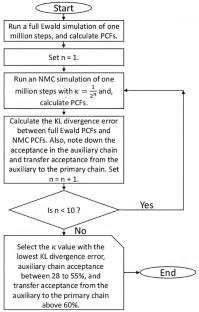
在这项工作中,针对对称和非对称离子系统进行了嵌套蒙特卡罗(NMC)模拟,其中能量函数由硬球和库仑相互作用(原始模型)组合描述。在 NMC 方法中,主链(能量函数为原始模型)的蒙特卡洛(MC)移动是通过使用辅助势能运行短 MC 轨迹来实现的,从而大大降低了计算成本。在我们的工作中,使用了 Debye-Hückel (DH) 势作为辅助势。结果表明,只要谨慎选择 DH 势中的 Debye 长度和辅助链中短 MC 运行的长度,NMC 方法就能得到与在原始模型中使用全 Ewald 求和法进行的更昂贵 MC 模拟相同的结果。通过最小化标准(无任何辅助势)MC 仿真与 NMC 的对相关函数之间的 Kullback-Leibler (KL) 分歧,还提出了一种简单算法,用于开发良好的 DH 类势,作为嵌套 MC 运行中的辅助势。总之,这项技术大大降低了埃瓦尔德求和技术的计算成本,并可应用于势能函数中存在库仑相互作用的任何原子和分子系统。图解摘要对对称和非对称离子系统进行了嵌套蒙特卡罗(NMC)模拟,其中主链的蒙特卡罗(MC)移动是通过使用辅助势能运行短的 MC 轨迹给出的。通过 NMC 计算出的离子对相关函数与标准的埃瓦尔德模拟结果非常吻合。
Nested Monte Carlo simulation of ionic systems with the primitive model using Debye-Hückel (DH) potential as an importance function and optimizing the DH potential with Kullback-Leibler divergence minimization
In this work, Nested Monte Carlo (NMC) simulation was done for symmetric and asymmetric ionic systems where the energy function is described by a combination of hard-sphere and Coulomb interaction (the primitive model). In the NMC method, Monte Carlo (MC) moves for the primary chain (where the energy function is the primitive model) is given by running a short MC trajectory using an auxiliary potential reducing the computational cost significantly. The Debye-Hückel (DH) potential was used as the auxiliary potential in our work. It is shown that with a careful choice of Debye length in the DH potential, and length of the short MC run in the auxiliary chain, the NMC method gives the same result as the more expensive MC simulation using full Ewald summation in the primitive model. Implementing the minimization of the Kullback–Leibler (KL) divergence between the pair-correlation function of the standard (without any auxiliary potential) MC simulation and NMC, a simple algorithm was also presented to develop good DH-like potentials to be used as auxiliary potentials in the nested MC run. Overall, this technique significantly reduces the Ewald summation technique's computational cost and can be applied to any atomic and molecular system with coulombic interaction in the potential energy function.
Graphical abstract
Nested Monte Carlo (NMC) simulations for symmetric and asymmetric ionic systems are performed, where Monte Carlo (MC) moves for the primary chain are given by running a short MC trajectory using an auxiliary potential. Excellent agreement has been shown between the pair correlation functions calculated from NMC, and standard Ewald simulations.
期刊介绍:
Journal of Chemical Sciences is a monthly journal published by the Indian Academy of Sciences. It formed part of the original Proceedings of the Indian Academy of Sciences – Part A, started by the Nobel Laureate Prof C V Raman in 1934, that was split in 1978 into three separate journals. It was renamed as Journal of Chemical Sciences in 2004. The journal publishes original research articles and rapid communications, covering all areas of chemical sciences. A significant feature of the journal is its special issues, brought out from time to time, devoted to conference symposia/proceedings in frontier areas of the subject, held not only in India but also in other countries.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
