Edoardo Giuili, Robin Grolaux, Catarina Z N M Macedo, Laurence Desmyter, Bruno Pichon, Sebastian Neuens, Catheline Vilain, Catharina Olsen, Sonia Van Dooren, Guillaume Smits, Matthieu Defrance
{"title":"用于神经发育障碍(NDD)诊断的附加信号实施的综合评估。","authors":"Edoardo Giuili, Robin Grolaux, Catarina Z N M Macedo, Laurence Desmyter, Bruno Pichon, Sebastian Neuens, Catheline Vilain, Catharina Olsen, Sonia Van Dooren, Guillaume Smits, Matthieu Defrance","doi":"10.1007/s00439-023-02609-2","DOIUrl":null,"url":null,"abstract":"<p><p>Episignatures are popular tools for the diagnosis of rare neurodevelopmental disorders. They are commonly based on a set of differentially methylated CpGs used in combination with a support vector machine model. DNA methylation (DNAm) data often include missing values due to changes in data generation technology and batch effects. While many normalization methods exist for DNAm data, their impact on episignature performance have never been assessed. In addition, technologies to quantify DNAm evolve quickly and this may lead to poor transposition of existing episignatures generated on deprecated array versions to new ones. Indeed, probe removal between array versions, technologies or during preprocessing leads to missing values. Thus, the effect of missing data on episignature performance must also be carefully evaluated and addressed through imputation or an innovative approach to episignatures design. In this paper, we used data from patients suffering from Kabuki and Sotos syndrome to evaluate the influence of normalization methods, classification models and missing data on the prediction performances of two existing episignatures. We compare how six popular normalization methods for methylarray data affect episignature classification performances in Kabuki and Sotos syndromes and provide best practice suggestions when building new episignatures. In this setting, we show that Illumina, Noob or Funnorm normalization methods achieved higher classification performances on the testing sets compared to Quantile, Raw and Swan normalization methods. We further show that penalized logistic regression and support vector machines perform best in the classification of Kabuki and Sotos syndrome patients. Then, we describe a new paradigm to build episignatures based on the detection of differentially methylated regions (DMRs) and evaluate their performance compared to classical differentially methylated cytosines (DMCs)-based episignatures in the presence of missing data. We show that the performance of classical DMC-based episignatures suffers from the presence of missing data more than the DMR-based approach. We present a comprehensive evaluation of how the normalization of DNA methylation data affects episignature performance, using three popular classification models. We further evaluate how missing data affect those models' predictions. Finally, we propose a novel methodology to develop episignatures based on differentially methylated regions identification and show how this method slightly outperforms classical episignatures in the presence of missing data.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":" ","pages":"1721-1735"},"PeriodicalIF":3.6000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10676303/pdf/","citationCount":"0","resultStr":"{\"title\":\"Comprehensive evaluation of the implementation of episignatures for diagnosis of neurodevelopmental disorders (NDDs).\",\"authors\":\"Edoardo Giuili, Robin Grolaux, Catarina Z N M Macedo, Laurence Desmyter, Bruno Pichon, Sebastian Neuens, Catheline Vilain, Catharina Olsen, Sonia Van Dooren, Guillaume Smits, Matthieu Defrance\",\"doi\":\"10.1007/s00439-023-02609-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Episignatures are popular tools for the diagnosis of rare neurodevelopmental disorders. They are commonly based on a set of differentially methylated CpGs used in combination with a support vector machine model. DNA methylation (DNAm) data often include missing values due to changes in data generation technology and batch effects. While many normalization methods exist for DNAm data, their impact on episignature performance have never been assessed. In addition, technologies to quantify DNAm evolve quickly and this may lead to poor transposition of existing episignatures generated on deprecated array versions to new ones. Indeed, probe removal between array versions, technologies or during preprocessing leads to missing values. Thus, the effect of missing data on episignature performance must also be carefully evaluated and addressed through imputation or an innovative approach to episignatures design. In this paper, we used data from patients suffering from Kabuki and Sotos syndrome to evaluate the influence of normalization methods, classification models and missing data on the prediction performances of two existing episignatures. We compare how six popular normalization methods for methylarray data affect episignature classification performances in Kabuki and Sotos syndromes and provide best practice suggestions when building new episignatures. In this setting, we show that Illumina, Noob or Funnorm normalization methods achieved higher classification performances on the testing sets compared to Quantile, Raw and Swan normalization methods. We further show that penalized logistic regression and support vector machines perform best in the classification of Kabuki and Sotos syndrome patients. Then, we describe a new paradigm to build episignatures based on the detection of differentially methylated regions (DMRs) and evaluate their performance compared to classical differentially methylated cytosines (DMCs)-based episignatures in the presence of missing data. We show that the performance of classical DMC-based episignatures suffers from the presence of missing data more than the DMR-based approach. We present a comprehensive evaluation of how the normalization of DNA methylation data affects episignature performance, using three popular classification models. We further evaluate how missing data affect those models' predictions. Finally, we propose a novel methodology to develop episignatures based on differentially methylated regions identification and show how this method slightly outperforms classical episignatures in the presence of missing data.</p>\",\"PeriodicalId\":13175,\"journal\":{\"name\":\"Human Genetics\",\"volume\":\" \",\"pages\":\"1721-1735\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2023-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10676303/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s00439-023-02609-2\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/10/27 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-023-02609-2","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/10/27 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Comprehensive evaluation of the implementation of episignatures for diagnosis of neurodevelopmental disorders (NDDs).
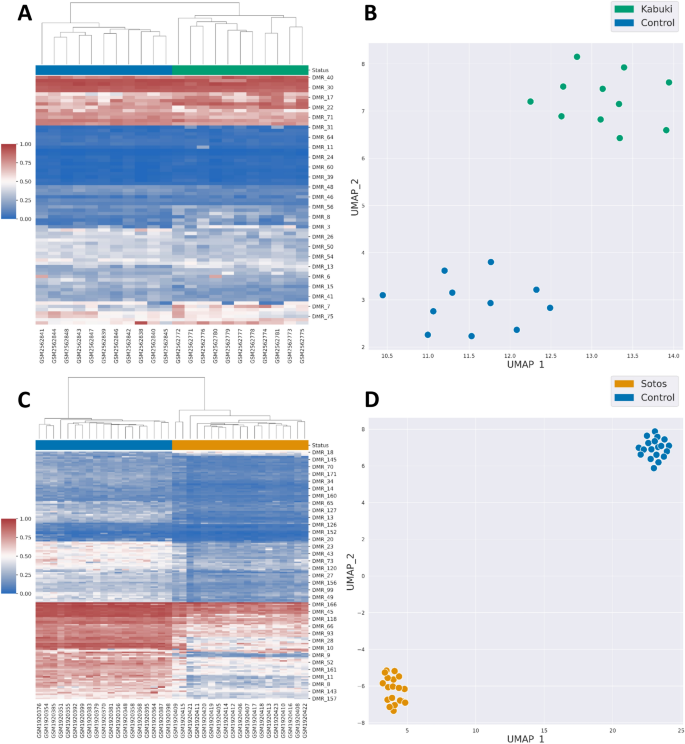
Episignatures are popular tools for the diagnosis of rare neurodevelopmental disorders. They are commonly based on a set of differentially methylated CpGs used in combination with a support vector machine model. DNA methylation (DNAm) data often include missing values due to changes in data generation technology and batch effects. While many normalization methods exist for DNAm data, their impact on episignature performance have never been assessed. In addition, technologies to quantify DNAm evolve quickly and this may lead to poor transposition of existing episignatures generated on deprecated array versions to new ones. Indeed, probe removal between array versions, technologies or during preprocessing leads to missing values. Thus, the effect of missing data on episignature performance must also be carefully evaluated and addressed through imputation or an innovative approach to episignatures design. In this paper, we used data from patients suffering from Kabuki and Sotos syndrome to evaluate the influence of normalization methods, classification models and missing data on the prediction performances of two existing episignatures. We compare how six popular normalization methods for methylarray data affect episignature classification performances in Kabuki and Sotos syndromes and provide best practice suggestions when building new episignatures. In this setting, we show that Illumina, Noob or Funnorm normalization methods achieved higher classification performances on the testing sets compared to Quantile, Raw and Swan normalization methods. We further show that penalized logistic regression and support vector machines perform best in the classification of Kabuki and Sotos syndrome patients. Then, we describe a new paradigm to build episignatures based on the detection of differentially methylated regions (DMRs) and evaluate their performance compared to classical differentially methylated cytosines (DMCs)-based episignatures in the presence of missing data. We show that the performance of classical DMC-based episignatures suffers from the presence of missing data more than the DMR-based approach. We present a comprehensive evaluation of how the normalization of DNA methylation data affects episignature performance, using three popular classification models. We further evaluate how missing data affect those models' predictions. Finally, we propose a novel methodology to develop episignatures based on differentially methylated regions identification and show how this method slightly outperforms classical episignatures in the presence of missing data.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
