{"title":"高压下液态Fe-Ni-C体系的成分效应","authors":"Esther S. Posner, Gerd Steinle-Neumann","doi":"10.1007/s00269-022-01219-0","DOIUrl":null,"url":null,"abstract":"<div><p>We performed molecular dynamics simulations based on density functional theory to systematically investigate the Fe–Ni–C system including (1) pure Fe and Ni; (2) binary Fe–Ni, Fe–C, and Ni–C; and (3) ternary Fe–Ni–C liquid compositions at 3000 K and three simulation volumes corresponding to pressure (<i>P</i>) up to 83 GPa. Liquid structural properties, including coordination numbers, are analyzed using partial radial distribution functions. Self-diffusion coefficients are determined based on the atomic trajectories and the asymptotic slope of the time-dependent mean-square displacement. The results indicate that the average interatomic distance between two Fe atoms (<i>r</i><sub>Fe–Fe</sub>) decreases with <i>P</i> and is sensitive to Ni (<i>X</i><sub>Ni</sub>) and C (<i>X</i><sub>C</sub>) concentration, although the effects are opposite: <i>r</i><sub>Fe–Fe</sub> decreases with increasing <i>X</i><sub>Ni</sub>, but increases with increasing <i>X</i><sub>C</sub>. Average <i>r</i><sub>Fe–C</sub> and <i>r</i><sub>Ni–C</sub> values also decrease with increasing <i>X</i><sub>Ni</sub> and generally remain constant between the two lowest <i>P</i> points, corresponding to a coordination change of carbon from ~ 6.8 to ~ 8.0, and then decrease with additional <i>P</i> once the coordination change is complete. Carbon clustering occurs in both binary (especially Ni–C) and ternary compositions with short-range <i>r</i><sub>C-C</sub> values (~ 1.29 to ~ 1.57 Å), typical for <i>r</i><sub>C-C</sub> in diamond and graphite. The self-diffusion results are generally consistent with high-<i>P</i> diffusion data extrapolated from experiments conducted at lower temperature (<i>T</i>). A subset of additional simulations was conducted at 1675 and 2350 K to estimate the effect of <i>T</i> on diffusion, yielding an activation enthalpy of ~ 53 kJ/mol and activation volume of ~ 0.5 cm<sup>3</sup>/mol.\n</p></div>","PeriodicalId":20132,"journal":{"name":"Physics and Chemistry of Minerals","volume":"49 11","pages":""},"PeriodicalIF":1.6000,"publicationDate":"2022-10-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s00269-022-01219-0.pdf","citationCount":"0","resultStr":"{\"title\":\"Compositional effects in the liquid Fe–Ni–C system at high pressure\",\"authors\":\"Esther S. Posner, Gerd Steinle-Neumann\",\"doi\":\"10.1007/s00269-022-01219-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>We performed molecular dynamics simulations based on density functional theory to systematically investigate the Fe–Ni–C system including (1) pure Fe and Ni; (2) binary Fe–Ni, Fe–C, and Ni–C; and (3) ternary Fe–Ni–C liquid compositions at 3000 K and three simulation volumes corresponding to pressure (<i>P</i>) up to 83 GPa. Liquid structural properties, including coordination numbers, are analyzed using partial radial distribution functions. Self-diffusion coefficients are determined based on the atomic trajectories and the asymptotic slope of the time-dependent mean-square displacement. The results indicate that the average interatomic distance between two Fe atoms (<i>r</i><sub>Fe–Fe</sub>) decreases with <i>P</i> and is sensitive to Ni (<i>X</i><sub>Ni</sub>) and C (<i>X</i><sub>C</sub>) concentration, although the effects are opposite: <i>r</i><sub>Fe–Fe</sub> decreases with increasing <i>X</i><sub>Ni</sub>, but increases with increasing <i>X</i><sub>C</sub>. Average <i>r</i><sub>Fe–C</sub> and <i>r</i><sub>Ni–C</sub> values also decrease with increasing <i>X</i><sub>Ni</sub> and generally remain constant between the two lowest <i>P</i> points, corresponding to a coordination change of carbon from ~ 6.8 to ~ 8.0, and then decrease with additional <i>P</i> once the coordination change is complete. Carbon clustering occurs in both binary (especially Ni–C) and ternary compositions with short-range <i>r</i><sub>C-C</sub> values (~ 1.29 to ~ 1.57 Å), typical for <i>r</i><sub>C-C</sub> in diamond and graphite. The self-diffusion results are generally consistent with high-<i>P</i> diffusion data extrapolated from experiments conducted at lower temperature (<i>T</i>). A subset of additional simulations was conducted at 1675 and 2350 K to estimate the effect of <i>T</i> on diffusion, yielding an activation enthalpy of ~ 53 kJ/mol and activation volume of ~ 0.5 cm<sup>3</sup>/mol.\\n</p></div>\",\"PeriodicalId\":20132,\"journal\":{\"name\":\"Physics and Chemistry of Minerals\",\"volume\":\"49 11\",\"pages\":\"\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2022-10-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://link.springer.com/content/pdf/10.1007/s00269-022-01219-0.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physics and Chemistry of Minerals\",\"FirstCategoryId\":\"89\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00269-022-01219-0\",\"RegionNum\":4,\"RegionCategory\":\"地球科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"MATERIALS SCIENCE, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physics and Chemistry of Minerals","FirstCategoryId":"89","ListUrlMain":"https://link.springer.com/article/10.1007/s00269-022-01219-0","RegionNum":4,"RegionCategory":"地球科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
Compositional effects in the liquid Fe–Ni–C system at high pressure
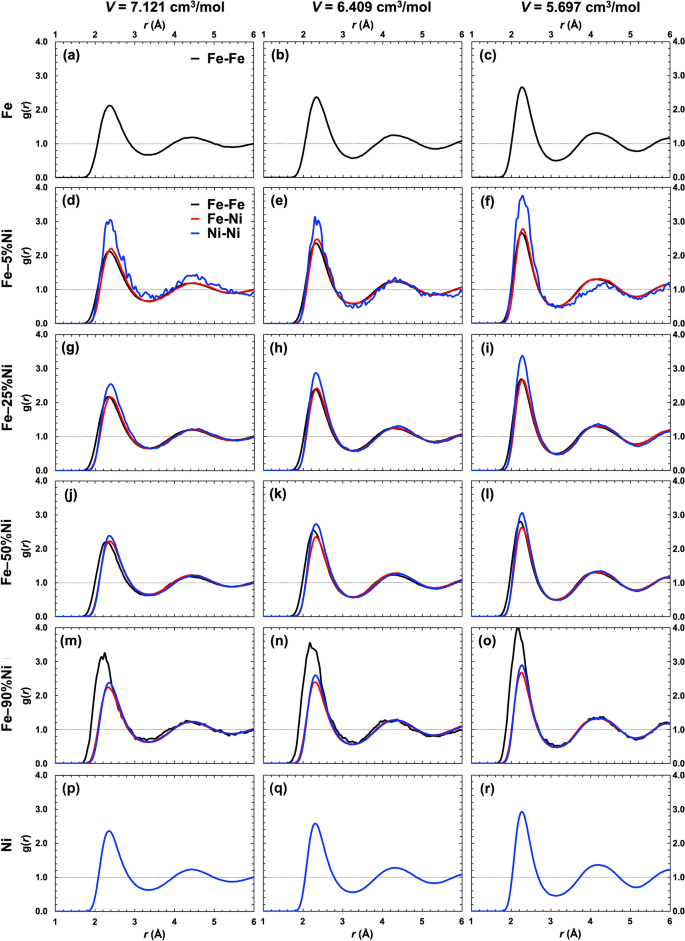
We performed molecular dynamics simulations based on density functional theory to systematically investigate the Fe–Ni–C system including (1) pure Fe and Ni; (2) binary Fe–Ni, Fe–C, and Ni–C; and (3) ternary Fe–Ni–C liquid compositions at 3000 K and three simulation volumes corresponding to pressure (P) up to 83 GPa. Liquid structural properties, including coordination numbers, are analyzed using partial radial distribution functions. Self-diffusion coefficients are determined based on the atomic trajectories and the asymptotic slope of the time-dependent mean-square displacement. The results indicate that the average interatomic distance between two Fe atoms (rFe–Fe) decreases with P and is sensitive to Ni (XNi) and C (XC) concentration, although the effects are opposite: rFe–Fe decreases with increasing XNi, but increases with increasing XC. Average rFe–C and rNi–C values also decrease with increasing XNi and generally remain constant between the two lowest P points, corresponding to a coordination change of carbon from ~ 6.8 to ~ 8.0, and then decrease with additional P once the coordination change is complete. Carbon clustering occurs in both binary (especially Ni–C) and ternary compositions with short-range rC-C values (~ 1.29 to ~ 1.57 Å), typical for rC-C in diamond and graphite. The self-diffusion results are generally consistent with high-P diffusion data extrapolated from experiments conducted at lower temperature (T). A subset of additional simulations was conducted at 1675 and 2350 K to estimate the effect of T on diffusion, yielding an activation enthalpy of ~ 53 kJ/mol and activation volume of ~ 0.5 cm3/mol.
期刊介绍:
Physics and Chemistry of Minerals is an international journal devoted to publishing articles and short communications of physical or chemical studies on minerals or solids related to minerals. The aim of the journal is to support competent interdisciplinary work in mineralogy and physics or chemistry. Particular emphasis is placed on applications of modern techniques or new theories and models to interpret atomic structures and physical or chemical properties of minerals. Some subjects of interest are:
-Relationships between atomic structure and crystalline state (structures of various states, crystal energies, crystal growth, thermodynamic studies, phase transformations, solid solution, exsolution phenomena, etc.)
-General solid state spectroscopy (ultraviolet, visible, infrared, Raman, ESCA, luminescence, X-ray, electron paramagnetic resonance, nuclear magnetic resonance, gamma ray resonance, etc.)
-Experimental and theoretical analysis of chemical bonding in minerals (application of crystal field, molecular orbital, band theories, etc.)
-Physical properties (magnetic, mechanical, electric, optical, thermodynamic, etc.)
-Relations between thermal expansion, compressibility, elastic constants, and fundamental properties of atomic structure, particularly as applied to geophysical problems
-Electron microscopy in support of physical and chemical studies
-Computational methods in the study of the structure and properties of minerals
-Mineral surfaces (experimental methods, structure and properties)



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
