Youjin Xiong, Yiqing Wang, Yisheng Wang, Chenmei Li, Peng Yusong, Junyu Wu, Yiqing Wang, Lingyun Gu, Christopher J. Butch
{"title":"利用基于贝叶斯对接近似训练的强化学习的混合深度生成模型改进药物发现","authors":"Youjin Xiong, Yiqing Wang, Yisheng Wang, Chenmei Li, Peng Yusong, Junyu Wu, Yiqing Wang, Lingyun Gu, Christopher J. Butch","doi":"10.1007/s10822-023-00523-3","DOIUrl":null,"url":null,"abstract":"<div><p>Generative approaches to molecular design are an area of intense study in recent years as a method to generate new pharmaceuticals with desired properties. Often though, these types of efforts are constrained by limited experimental activity data, resulting in either models that generate molecules with poor performance or models that are overfit and produce close analogs of known molecules. In this paper, we reduce this data dependency for the generation of new chemotypes by incorporating docking scores of known and de novo molecules to expand the applicability domain of the reward function and diversify the compounds generated during reinforcement learning. Our approach employs a deep generative model initially trained using a combination of limited known drug activity and an approximate docking score provided by a second machine learned Bayes regression model, with final evaluation of high scoring compounds by a full docking simulation. This strategy results in molecules with docking scores improved by 10–20% compared to molecules of similar size, while being 130 × faster than a docking only approach on a typical GPU workstation. We also show that the increased docking scores correlate with (1) docking poses with interactions similar to known inhibitors and (2) result in higher MM-GBSA binding energies comparable to the energies of known DDR1 inhibitors, demonstrating that the Bayesian model contains sufficient information for the network to learn to efficiently interact with the binding pocket during reinforcement learning. This outcome shows that the combination of the learned latent molecular representation along with the feature-based docking regression is sufficient for reinforcement learning to infer the relationship between the molecules and the receptor binding site, which suggest that our method can be a powerful tool for the discovery of new chemotypes with potential therapeutic applications.</p></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"37 11","pages":"507 - 517"},"PeriodicalIF":3.1000,"publicationDate":"2023-08-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s10822-023-00523-3.pdf","citationCount":"0","resultStr":"{\"title\":\"Improving drug discovery with a hybrid deep generative model using reinforcement learning trained on a Bayesian docking approximation\",\"authors\":\"Youjin Xiong, Yiqing Wang, Yisheng Wang, Chenmei Li, Peng Yusong, Junyu Wu, Yiqing Wang, Lingyun Gu, Christopher J. Butch\",\"doi\":\"10.1007/s10822-023-00523-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Generative approaches to molecular design are an area of intense study in recent years as a method to generate new pharmaceuticals with desired properties. Often though, these types of efforts are constrained by limited experimental activity data, resulting in either models that generate molecules with poor performance or models that are overfit and produce close analogs of known molecules. In this paper, we reduce this data dependency for the generation of new chemotypes by incorporating docking scores of known and de novo molecules to expand the applicability domain of the reward function and diversify the compounds generated during reinforcement learning. Our approach employs a deep generative model initially trained using a combination of limited known drug activity and an approximate docking score provided by a second machine learned Bayes regression model, with final evaluation of high scoring compounds by a full docking simulation. This strategy results in molecules with docking scores improved by 10–20% compared to molecules of similar size, while being 130 × faster than a docking only approach on a typical GPU workstation. We also show that the increased docking scores correlate with (1) docking poses with interactions similar to known inhibitors and (2) result in higher MM-GBSA binding energies comparable to the energies of known DDR1 inhibitors, demonstrating that the Bayesian model contains sufficient information for the network to learn to efficiently interact with the binding pocket during reinforcement learning. This outcome shows that the combination of the learned latent molecular representation along with the feature-based docking regression is sufficient for reinforcement learning to infer the relationship between the molecules and the receptor binding site, which suggest that our method can be a powerful tool for the discovery of new chemotypes with potential therapeutic applications.</p></div>\",\"PeriodicalId\":621,\"journal\":{\"name\":\"Journal of Computer-Aided Molecular Design\",\"volume\":\"37 11\",\"pages\":\"507 - 517\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2023-08-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://link.springer.com/content/pdf/10.1007/s10822-023-00523-3.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computer-Aided Molecular Design\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10822-023-00523-3\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-023-00523-3","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Improving drug discovery with a hybrid deep generative model using reinforcement learning trained on a Bayesian docking approximation
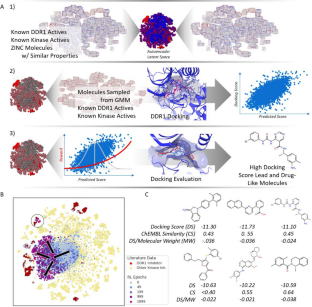
Generative approaches to molecular design are an area of intense study in recent years as a method to generate new pharmaceuticals with desired properties. Often though, these types of efforts are constrained by limited experimental activity data, resulting in either models that generate molecules with poor performance or models that are overfit and produce close analogs of known molecules. In this paper, we reduce this data dependency for the generation of new chemotypes by incorporating docking scores of known and de novo molecules to expand the applicability domain of the reward function and diversify the compounds generated during reinforcement learning. Our approach employs a deep generative model initially trained using a combination of limited known drug activity and an approximate docking score provided by a second machine learned Bayes regression model, with final evaluation of high scoring compounds by a full docking simulation. This strategy results in molecules with docking scores improved by 10–20% compared to molecules of similar size, while being 130 × faster than a docking only approach on a typical GPU workstation. We also show that the increased docking scores correlate with (1) docking poses with interactions similar to known inhibitors and (2) result in higher MM-GBSA binding energies comparable to the energies of known DDR1 inhibitors, demonstrating that the Bayesian model contains sufficient information for the network to learn to efficiently interact with the binding pocket during reinforcement learning. This outcome shows that the combination of the learned latent molecular representation along with the feature-based docking regression is sufficient for reinforcement learning to infer the relationship between the molecules and the receptor binding site, which suggest that our method can be a powerful tool for the discovery of new chemotypes with potential therapeutic applications.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
