Lucrezia Catapano, Fei Long, Keitaro Yamashita, Robert A Nicholls, Roberto A Steiner, Garib N Murshudov
{"title":"利用CCP4套件中的REFMAC5进行中子晶体学精炼。","authors":"Lucrezia Catapano, Fei Long, Keitaro Yamashita, Robert A Nicholls, Roberto A Steiner, Garib N Murshudov","doi":"10.1107/S2059798323008793","DOIUrl":null,"url":null,"abstract":"<p><p>Hydrogen (H) atoms are abundant in macromolecules and often play critical roles in enzyme catalysis, ligand-recognition processes and protein-protein interactions. However, their direct visualization by diffraction techniques is challenging. Macromolecular X-ray crystallography affords the localization of only the most ordered H atoms at (sub-)atomic resolution (around 1.2 Å or higher). However, many H atoms of biochemical significance remain undetectable by this method. In contrast, neutron diffraction methods enable the visualization of most H atoms, typically in the form of deuterium (<sup>2</sup>H) atoms, at much more common resolution values (better than 2.5 Å). Thus, neutron crystallography, although technically demanding, is often the method of choice when direct information on protonation states is sought. REFMAC5 from the Collaborative Computational Project No. 4 (CCP4) is a program for the refinement of macromolecular models against X-ray crystallographic and cryo-EM data. This contribution describes its extension to include the refinement of structural models obtained from neutron crystallographic data. Stereochemical restraints with accurate bond distances between H atoms and their parent atom nuclei are now part of the CCP4 Monomer Library, the source of prior chemical information used in the refinement. One new feature for neutron data analysis in REFMAC5 is refinement of the protium/deuterium (<sup>1</sup>H/<sup>2</sup>H) fraction. This parameter describes the relative <sup>1</sup>H/<sup>2</sup>H contribution to neutron scattering for hydrogen isotopes. The newly developed REFMAC5 algorithms were tested by performing the (re-)refinement of several entries available in the PDB and of one novel structure (FutA) using either (i) neutron data only or (ii) neutron data supplemented by external restraints to a reference X-ray crystallographic structure. Re-refinement with REFMAC5 afforded models characterized by R-factor values that are consistent with, and in some cases better than, the originally deposited values. The use of external reference structure restraints during refinement has been observed to be a valuable strategy, especially for structures at medium-low resolution.</p>","PeriodicalId":7116,"journal":{"name":"Acta Crystallographica. Section D, Structural Biology","volume":" ","pages":"1056-1070"},"PeriodicalIF":3.8000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7615533/pdf/","citationCount":"0","resultStr":"{\"title\":\"Neutron crystallographic refinement with REFMAC5 from the CCP4 suite.\",\"authors\":\"Lucrezia Catapano, Fei Long, Keitaro Yamashita, Robert A Nicholls, Roberto A Steiner, Garib N Murshudov\",\"doi\":\"10.1107/S2059798323008793\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Hydrogen (H) atoms are abundant in macromolecules and often play critical roles in enzyme catalysis, ligand-recognition processes and protein-protein interactions. However, their direct visualization by diffraction techniques is challenging. Macromolecular X-ray crystallography affords the localization of only the most ordered H atoms at (sub-)atomic resolution (around 1.2 Å or higher). However, many H atoms of biochemical significance remain undetectable by this method. In contrast, neutron diffraction methods enable the visualization of most H atoms, typically in the form of deuterium (<sup>2</sup>H) atoms, at much more common resolution values (better than 2.5 Å). Thus, neutron crystallography, although technically demanding, is often the method of choice when direct information on protonation states is sought. REFMAC5 from the Collaborative Computational Project No. 4 (CCP4) is a program for the refinement of macromolecular models against X-ray crystallographic and cryo-EM data. This contribution describes its extension to include the refinement of structural models obtained from neutron crystallographic data. Stereochemical restraints with accurate bond distances between H atoms and their parent atom nuclei are now part of the CCP4 Monomer Library, the source of prior chemical information used in the refinement. One new feature for neutron data analysis in REFMAC5 is refinement of the protium/deuterium (<sup>1</sup>H/<sup>2</sup>H) fraction. This parameter describes the relative <sup>1</sup>H/<sup>2</sup>H contribution to neutron scattering for hydrogen isotopes. The newly developed REFMAC5 algorithms were tested by performing the (re-)refinement of several entries available in the PDB and of one novel structure (FutA) using either (i) neutron data only or (ii) neutron data supplemented by external restraints to a reference X-ray crystallographic structure. Re-refinement with REFMAC5 afforded models characterized by R-factor values that are consistent with, and in some cases better than, the originally deposited values. The use of external reference structure restraints during refinement has been observed to be a valuable strategy, especially for structures at medium-low resolution.</p>\",\"PeriodicalId\":7116,\"journal\":{\"name\":\"Acta Crystallographica. Section D, Structural Biology\",\"volume\":\" \",\"pages\":\"1056-1070\"},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2023-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7615533/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Acta Crystallographica. Section D, Structural Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1107/S2059798323008793\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/11/3 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Crystallographica. Section D, Structural Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1107/S2059798323008793","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/11/3 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Neutron crystallographic refinement with REFMAC5 from the CCP4 suite.
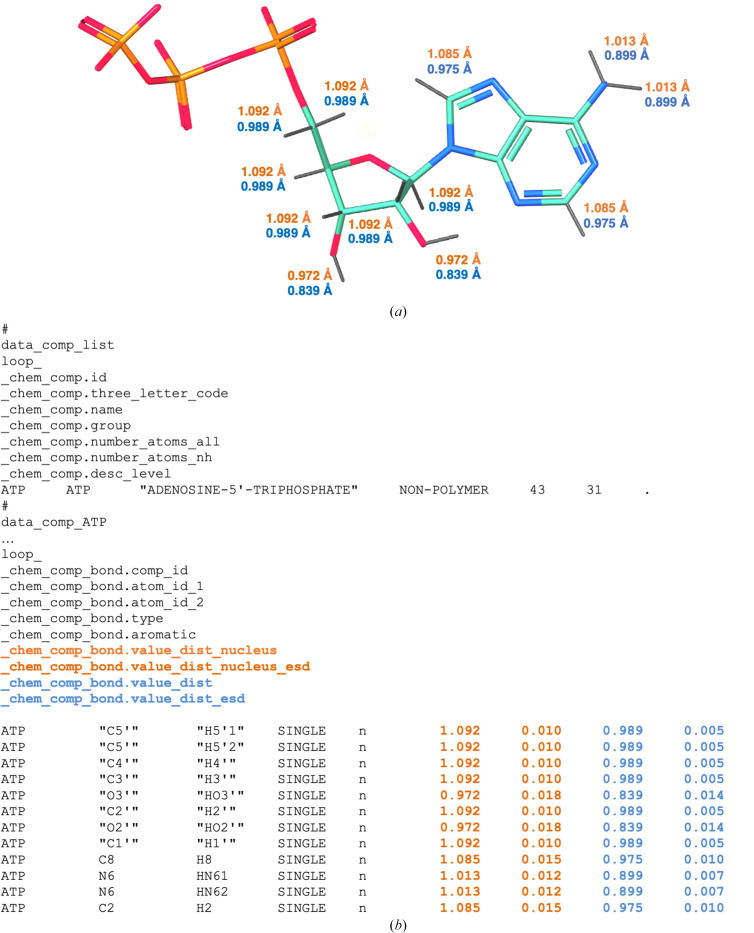
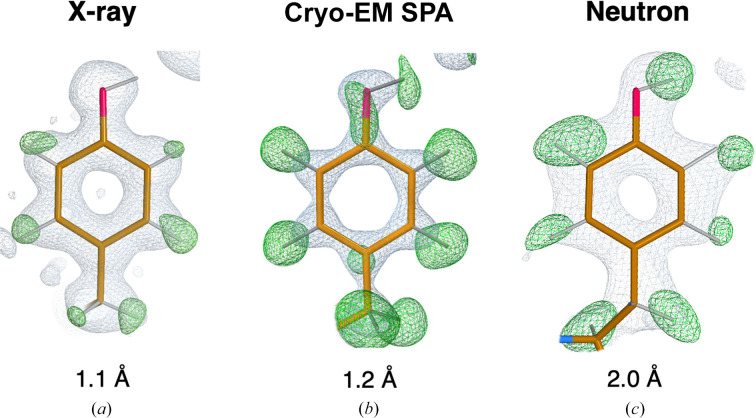
Hydrogen (H) atoms are abundant in macromolecules and often play critical roles in enzyme catalysis, ligand-recognition processes and protein-protein interactions. However, their direct visualization by diffraction techniques is challenging. Macromolecular X-ray crystallography affords the localization of only the most ordered H atoms at (sub-)atomic resolution (around 1.2 Å or higher). However, many H atoms of biochemical significance remain undetectable by this method. In contrast, neutron diffraction methods enable the visualization of most H atoms, typically in the form of deuterium (2H) atoms, at much more common resolution values (better than 2.5 Å). Thus, neutron crystallography, although technically demanding, is often the method of choice when direct information on protonation states is sought. REFMAC5 from the Collaborative Computational Project No. 4 (CCP4) is a program for the refinement of macromolecular models against X-ray crystallographic and cryo-EM data. This contribution describes its extension to include the refinement of structural models obtained from neutron crystallographic data. Stereochemical restraints with accurate bond distances between H atoms and their parent atom nuclei are now part of the CCP4 Monomer Library, the source of prior chemical information used in the refinement. One new feature for neutron data analysis in REFMAC5 is refinement of the protium/deuterium (1H/2H) fraction. This parameter describes the relative 1H/2H contribution to neutron scattering for hydrogen isotopes. The newly developed REFMAC5 algorithms were tested by performing the (re-)refinement of several entries available in the PDB and of one novel structure (FutA) using either (i) neutron data only or (ii) neutron data supplemented by external restraints to a reference X-ray crystallographic structure. Re-refinement with REFMAC5 afforded models characterized by R-factor values that are consistent with, and in some cases better than, the originally deposited values. The use of external reference structure restraints during refinement has been observed to be a valuable strategy, especially for structures at medium-low resolution.
期刊介绍:
Acta Crystallographica Section D welcomes the submission of articles covering any aspect of structural biology, with a particular emphasis on the structures of biological macromolecules or the methods used to determine them.
Reports on new structures of biological importance may address the smallest macromolecules to the largest complex molecular machines. These structures may have been determined using any structural biology technique including crystallography, NMR, cryoEM and/or other techniques. The key criterion is that such articles must present significant new insights into biological, chemical or medical sciences. The inclusion of complementary data that support the conclusions drawn from the structural studies (such as binding studies, mass spectrometry, enzyme assays, or analysis of mutants or other modified forms of biological macromolecule) is encouraged.
Methods articles may include new approaches to any aspect of biological structure determination or structure analysis but will only be accepted where they focus on new methods that are demonstrated to be of general applicability and importance to structural biology. Articles describing particularly difficult problems in structural biology are also welcomed, if the analysis would provide useful insights to others facing similar problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
