Harriet Risby , Guy Robinson , Nastassya Chandra , Grace King , Roberto Vivancos , Robert Smith , Daniel Thomas , Andrew Fox , Noel McCarthy , Rachel M. Chalmers
{"title":"应用一种新的多位点可变数串联重复分析(MLVA)方案对2022年春季威尔士和英格兰西北部细小隐孢子虫病例进行季节性调查","authors":"Harriet Risby , Guy Robinson , Nastassya Chandra , Grace King , Roberto Vivancos , Robert Smith , Daniel Thomas , Andrew Fox , Noel McCarthy , Rachel M. Chalmers","doi":"10.1016/j.crpvbd.2023.100151","DOIUrl":null,"url":null,"abstract":"<div><p>The protozoan <em>Cryptosporidium parvum</em> is an important cause of gastroenteritis in humans and livestock, and cryptosporidiosis outbreaks are common. However, a multi-locus genotyping scheme is not widely adopted. We describe the further development and application of a seven-locus multi-locus variable number of tandem repeats analysis (MLVA) scheme. From 28th March to 31st July 2022, confirmed <em>C. parvum</em> stools (<em>n</em> = 213) from cryptosporidiosis patients (cases) in Wales (<em>n</em> = 95) and the north west of England (<em>n</em> = 118) were tested by MLVA. Typability (defined as alleles identified at all seven loci in a sample) was 81.2% and discriminatory power estimated by Hunter Gaston Discriminatory Index was 0.99. A MLVA profile was constructed from the alleles, expressed in chromosomal order. Profiles were defined as simple (single allele at each locus) or mixed (more than one allele at any locus). A total of 161 MLVA profiles were identified; 13 were mixed, an additional 38 simple profiles contained null records, and 110 were complete simple profiles. A minimum spanning tree was constructed of simple MLVA profiles and those identical at all seven loci defined genetic clusters of cases (here, null records were considered as an allele); 77 cases formed 25 clusters, ranging from two to nine (mode = two) cases. The largest cluster, following epidemiological investigation, signalled a newly-identified outbreak. Two other cases with mixed profiles that contained the outbreak alleles were included in the outbreak investigation. In another epidemiologically-identified outbreak of six initial cases, MLVA detected two additional cases. In a third, small outbreak of three cases, identical MLVA profiles strengthened the microbiological evidence. Review of the performance characteristics of the individual loci and of the seven-locus scheme suggested that two loci might be candidates for review, but a larger dataset over a wider geographical area and longer timeframe will help inform decision-making about the scheme by user laboratories and stakeholders (such as public health agencies). This MLVA scheme is straightforward in use, fast and cheap compared to sequence-based methods, identifies mixed infections, provides an important tool for <em>C. parvum</em> surveillance, and can enhance outbreak investigations and public health action.</p></div>","PeriodicalId":94311,"journal":{"name":"Current research in parasitology & vector-borne diseases","volume":"4 ","pages":"Article 100151"},"PeriodicalIF":1.7000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2667114X23000390/pdfft?md5=4434a17c591dcde16d072e2f284155e9&pid=1-s2.0-S2667114X23000390-main.pdf","citationCount":"1","resultStr":"{\"title\":\"Application of a new multi-locus variable number tandem repeat analysis (MLVA) scheme for the seasonal investigation of Cryptosporidium parvum cases in Wales and the northwest of England, spring 2022\",\"authors\":\"Harriet Risby , Guy Robinson , Nastassya Chandra , Grace King , Roberto Vivancos , Robert Smith , Daniel Thomas , Andrew Fox , Noel McCarthy , Rachel M. Chalmers\",\"doi\":\"10.1016/j.crpvbd.2023.100151\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The protozoan <em>Cryptosporidium parvum</em> is an important cause of gastroenteritis in humans and livestock, and cryptosporidiosis outbreaks are common. However, a multi-locus genotyping scheme is not widely adopted. We describe the further development and application of a seven-locus multi-locus variable number of tandem repeats analysis (MLVA) scheme. From 28th March to 31st July 2022, confirmed <em>C. parvum</em> stools (<em>n</em> = 213) from cryptosporidiosis patients (cases) in Wales (<em>n</em> = 95) and the north west of England (<em>n</em> = 118) were tested by MLVA. Typability (defined as alleles identified at all seven loci in a sample) was 81.2% and discriminatory power estimated by Hunter Gaston Discriminatory Index was 0.99. A MLVA profile was constructed from the alleles, expressed in chromosomal order. Profiles were defined as simple (single allele at each locus) or mixed (more than one allele at any locus). A total of 161 MLVA profiles were identified; 13 were mixed, an additional 38 simple profiles contained null records, and 110 were complete simple profiles. A minimum spanning tree was constructed of simple MLVA profiles and those identical at all seven loci defined genetic clusters of cases (here, null records were considered as an allele); 77 cases formed 25 clusters, ranging from two to nine (mode = two) cases. The largest cluster, following epidemiological investigation, signalled a newly-identified outbreak. Two other cases with mixed profiles that contained the outbreak alleles were included in the outbreak investigation. In another epidemiologically-identified outbreak of six initial cases, MLVA detected two additional cases. In a third, small outbreak of three cases, identical MLVA profiles strengthened the microbiological evidence. Review of the performance characteristics of the individual loci and of the seven-locus scheme suggested that two loci might be candidates for review, but a larger dataset over a wider geographical area and longer timeframe will help inform decision-making about the scheme by user laboratories and stakeholders (such as public health agencies). This MLVA scheme is straightforward in use, fast and cheap compared to sequence-based methods, identifies mixed infections, provides an important tool for <em>C. parvum</em> surveillance, and can enhance outbreak investigations and public health action.</p></div>\",\"PeriodicalId\":94311,\"journal\":{\"name\":\"Current research in parasitology & vector-borne diseases\",\"volume\":\"4 \",\"pages\":\"Article 100151\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S2667114X23000390/pdfft?md5=4434a17c591dcde16d072e2f284155e9&pid=1-s2.0-S2667114X23000390-main.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Current research in parasitology & vector-borne diseases\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2667114X23000390\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"PARASITOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current research in parasitology & vector-borne diseases","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2667114X23000390","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"PARASITOLOGY","Score":null,"Total":0}
Application of a new multi-locus variable number tandem repeat analysis (MLVA) scheme for the seasonal investigation of Cryptosporidium parvum cases in Wales and the northwest of England, spring 2022
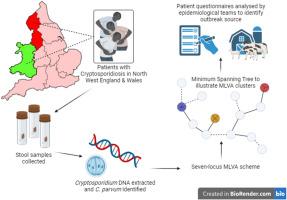
The protozoan Cryptosporidium parvum is an important cause of gastroenteritis in humans and livestock, and cryptosporidiosis outbreaks are common. However, a multi-locus genotyping scheme is not widely adopted. We describe the further development and application of a seven-locus multi-locus variable number of tandem repeats analysis (MLVA) scheme. From 28th March to 31st July 2022, confirmed C. parvum stools (n = 213) from cryptosporidiosis patients (cases) in Wales (n = 95) and the north west of England (n = 118) were tested by MLVA. Typability (defined as alleles identified at all seven loci in a sample) was 81.2% and discriminatory power estimated by Hunter Gaston Discriminatory Index was 0.99. A MLVA profile was constructed from the alleles, expressed in chromosomal order. Profiles were defined as simple (single allele at each locus) or mixed (more than one allele at any locus). A total of 161 MLVA profiles were identified; 13 were mixed, an additional 38 simple profiles contained null records, and 110 were complete simple profiles. A minimum spanning tree was constructed of simple MLVA profiles and those identical at all seven loci defined genetic clusters of cases (here, null records were considered as an allele); 77 cases formed 25 clusters, ranging from two to nine (mode = two) cases. The largest cluster, following epidemiological investigation, signalled a newly-identified outbreak. Two other cases with mixed profiles that contained the outbreak alleles were included in the outbreak investigation. In another epidemiologically-identified outbreak of six initial cases, MLVA detected two additional cases. In a third, small outbreak of three cases, identical MLVA profiles strengthened the microbiological evidence. Review of the performance characteristics of the individual loci and of the seven-locus scheme suggested that two loci might be candidates for review, but a larger dataset over a wider geographical area and longer timeframe will help inform decision-making about the scheme by user laboratories and stakeholders (such as public health agencies). This MLVA scheme is straightforward in use, fast and cheap compared to sequence-based methods, identifies mixed infections, provides an important tool for C. parvum surveillance, and can enhance outbreak investigations and public health action.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
