Xiangyue Zhao, Tingting Yu, Jie Tang, Ru-En Yao, Niu Li, Jian Wang
{"title":"2例KDM3B变异患者和新出现的Diets-Jongmans综合征。","authors":"Xiangyue Zhao, Tingting Yu, Jie Tang, Ru-En Yao, Niu Li, Jian Wang","doi":"10.1007/s10048-023-00711-1","DOIUrl":null,"url":null,"abstract":"<p><p>KDM3B is located on chromosome 5q31 and encodes KDM3B, which is involved in histone demethylation and epigenetic regulation. Pathogenic KDM3B variants cause a dominantly inherited disorder presenting with intellectual disability (ID), short stature, and facial dysmorphism, named Diets-Jongmans syndrome. We describe two patients with KDM3B variants presenting with Diets-Jongmans syndrome. Genetic testing was performed because of the clinical data and a lack of a clear diagnosis in both patients. Candidate variants were verified by Sanger sequencing. After KDM3B variants were detected, in silico tools were used to predict the pathogenicity of the missense variants. A minigene assay was performed to evaluate the splicing effects of the c.5070 + 1G > A variant on KDM3B. Patient 1 mainly presented with repetitive upper respiratory tract infection and patient 2 presented with palpitation, shortness of breath, and pitting edema; both had ID. Whole exome sequencing identified variants of KDM3B. Patient 1 had the de novo KDM3B c.5070 + 1G > A variant, whereas patient 2 had the c.2828G > A (p.R943Q) variant. Transcriptional experiments of the splicing variant c.5070 + 1G > A revealed aberrant transcripts leading to truncated protein products. We found two pathogenic variants in KDM3B, one of which is novel. Both patients had additional clinical presentations, and patient 1 had transient neutropenia. KDM3B c.5070 + 1G > A is the first KDM3B splice-site variant and was identified as a germline variant. Neutropenia and cardiomyopathy are newly found presentations of Diets-Jongmans syndrome. Our report enriches our knowledge of the genotypic spectrum of the KDM3B variants and phenotypic diversity of Diets-Jongmans syndrome.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":"24 2","pages":"95-101"},"PeriodicalIF":1.2000,"publicationDate":"2023-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":"{\"title\":\"Two patients with KDM3B variants and new presentations of Diets-Jongmans syndrome.\",\"authors\":\"Xiangyue Zhao, Tingting Yu, Jie Tang, Ru-En Yao, Niu Li, Jian Wang\",\"doi\":\"10.1007/s10048-023-00711-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>KDM3B is located on chromosome 5q31 and encodes KDM3B, which is involved in histone demethylation and epigenetic regulation. Pathogenic KDM3B variants cause a dominantly inherited disorder presenting with intellectual disability (ID), short stature, and facial dysmorphism, named Diets-Jongmans syndrome. We describe two patients with KDM3B variants presenting with Diets-Jongmans syndrome. Genetic testing was performed because of the clinical data and a lack of a clear diagnosis in both patients. Candidate variants were verified by Sanger sequencing. After KDM3B variants were detected, in silico tools were used to predict the pathogenicity of the missense variants. A minigene assay was performed to evaluate the splicing effects of the c.5070 + 1G > A variant on KDM3B. Patient 1 mainly presented with repetitive upper respiratory tract infection and patient 2 presented with palpitation, shortness of breath, and pitting edema; both had ID. Whole exome sequencing identified variants of KDM3B. Patient 1 had the de novo KDM3B c.5070 + 1G > A variant, whereas patient 2 had the c.2828G > A (p.R943Q) variant. Transcriptional experiments of the splicing variant c.5070 + 1G > A revealed aberrant transcripts leading to truncated protein products. We found two pathogenic variants in KDM3B, one of which is novel. Both patients had additional clinical presentations, and patient 1 had transient neutropenia. KDM3B c.5070 + 1G > A is the first KDM3B splice-site variant and was identified as a germline variant. Neutropenia and cardiomyopathy are newly found presentations of Diets-Jongmans syndrome. Our report enriches our knowledge of the genotypic spectrum of the KDM3B variants and phenotypic diversity of Diets-Jongmans syndrome.</p>\",\"PeriodicalId\":56106,\"journal\":{\"name\":\"Neurogenetics\",\"volume\":\"24 2\",\"pages\":\"95-101\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2023-04-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurogenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10048-023-00711-1\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-023-00711-1","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Two patients with KDM3B variants and new presentations of Diets-Jongmans syndrome.
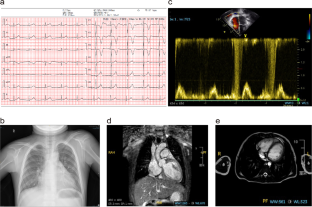
KDM3B is located on chromosome 5q31 and encodes KDM3B, which is involved in histone demethylation and epigenetic regulation. Pathogenic KDM3B variants cause a dominantly inherited disorder presenting with intellectual disability (ID), short stature, and facial dysmorphism, named Diets-Jongmans syndrome. We describe two patients with KDM3B variants presenting with Diets-Jongmans syndrome. Genetic testing was performed because of the clinical data and a lack of a clear diagnosis in both patients. Candidate variants were verified by Sanger sequencing. After KDM3B variants were detected, in silico tools were used to predict the pathogenicity of the missense variants. A minigene assay was performed to evaluate the splicing effects of the c.5070 + 1G > A variant on KDM3B. Patient 1 mainly presented with repetitive upper respiratory tract infection and patient 2 presented with palpitation, shortness of breath, and pitting edema; both had ID. Whole exome sequencing identified variants of KDM3B. Patient 1 had the de novo KDM3B c.5070 + 1G > A variant, whereas patient 2 had the c.2828G > A (p.R943Q) variant. Transcriptional experiments of the splicing variant c.5070 + 1G > A revealed aberrant transcripts leading to truncated protein products. We found two pathogenic variants in KDM3B, one of which is novel. Both patients had additional clinical presentations, and patient 1 had transient neutropenia. KDM3B c.5070 + 1G > A is the first KDM3B splice-site variant and was identified as a germline variant. Neutropenia and cardiomyopathy are newly found presentations of Diets-Jongmans syndrome. Our report enriches our knowledge of the genotypic spectrum of the KDM3B variants and phenotypic diversity of Diets-Jongmans syndrome.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
