Hanoch Senderowitz, Malkeet Singh Bahia, Omer Kaspi, Meir Touitou, Idan Binayev, Seema Dhail, Jacob Spiegel, Netaly Khazanov, Abraham Yosipof
{"title":"基于生物活性构象的QSAR建模中二维和三维描述符的比较。","authors":"Hanoch Senderowitz, Malkeet Singh Bahia, Omer Kaspi, Meir Touitou, Idan Binayev, Seema Dhail, Jacob Spiegel, Netaly Khazanov, Abraham Yosipof","doi":"10.1002/minf.202200186","DOIUrl":null,"url":null,"abstract":"<p><p>QSAR models are widely and successfully used in many research areas. The success of such models highly depends on molecular descriptors typically classified as 1D, 2D, 3D, or 4D. While 3D information is likely important, e. g., for modeling ligand-protein binding, previous comparisons between the performances of 2D and 3D descriptors were inconclusive. Yet in such comparisons the modeled ligands were not necessarily represented by their bioactive conformations. With this in mind, we mined the PDB for sets of protein-ligand complexes sharing the same protein for which uniform activity data were reported. The results, totaling 461 structures spread across six series were compiled into a carefully curated, first of its kind dataset in which each ligand is represented by its bioactive conformation. Next, each set was characterized by 2D, 3D and 2D + 3D descriptors and modeled using three machine learning algorithms, namely, k-Nearest Neighbors, Random Forest and Lasso Regression. Models' performances were evaluated on external test sets derived from the parent datasets either randomly or in a rational manner. We found that many more significant models were obtained when combining 2D and 3D descriptors. We attribute these improvements to the ability of 2D and 3D descriptors to code for different, yet complementary molecular properties.</p>","PeriodicalId":18853,"journal":{"name":"Molecular Informatics","volume":"42 4","pages":"e2200186"},"PeriodicalIF":3.1000,"publicationDate":"2023-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"2","resultStr":"{\"title\":\"A comparison between 2D and 3D descriptors in QSAR modeling based on bio-active conformations.\",\"authors\":\"Hanoch Senderowitz, Malkeet Singh Bahia, Omer Kaspi, Meir Touitou, Idan Binayev, Seema Dhail, Jacob Spiegel, Netaly Khazanov, Abraham Yosipof\",\"doi\":\"10.1002/minf.202200186\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>QSAR models are widely and successfully used in many research areas. The success of such models highly depends on molecular descriptors typically classified as 1D, 2D, 3D, or 4D. While 3D information is likely important, e. g., for modeling ligand-protein binding, previous comparisons between the performances of 2D and 3D descriptors were inconclusive. Yet in such comparisons the modeled ligands were not necessarily represented by their bioactive conformations. With this in mind, we mined the PDB for sets of protein-ligand complexes sharing the same protein for which uniform activity data were reported. The results, totaling 461 structures spread across six series were compiled into a carefully curated, first of its kind dataset in which each ligand is represented by its bioactive conformation. Next, each set was characterized by 2D, 3D and 2D + 3D descriptors and modeled using three machine learning algorithms, namely, k-Nearest Neighbors, Random Forest and Lasso Regression. Models' performances were evaluated on external test sets derived from the parent datasets either randomly or in a rational manner. We found that many more significant models were obtained when combining 2D and 3D descriptors. We attribute these improvements to the ability of 2D and 3D descriptors to code for different, yet complementary molecular properties.</p>\",\"PeriodicalId\":18853,\"journal\":{\"name\":\"Molecular Informatics\",\"volume\":\"42 4\",\"pages\":\"e2200186\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2023-04-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Informatics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/minf.202200186\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/minf.202200186","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
A comparison between 2D and 3D descriptors in QSAR modeling based on bio-active conformations.
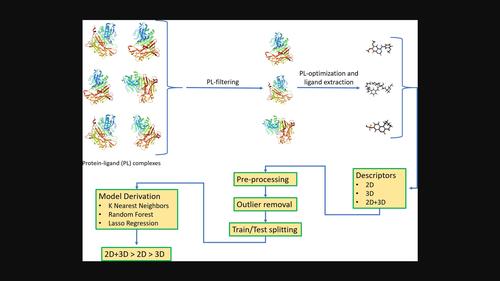
QSAR models are widely and successfully used in many research areas. The success of such models highly depends on molecular descriptors typically classified as 1D, 2D, 3D, or 4D. While 3D information is likely important, e. g., for modeling ligand-protein binding, previous comparisons between the performances of 2D and 3D descriptors were inconclusive. Yet in such comparisons the modeled ligands were not necessarily represented by their bioactive conformations. With this in mind, we mined the PDB for sets of protein-ligand complexes sharing the same protein for which uniform activity data were reported. The results, totaling 461 structures spread across six series were compiled into a carefully curated, first of its kind dataset in which each ligand is represented by its bioactive conformation. Next, each set was characterized by 2D, 3D and 2D + 3D descriptors and modeled using three machine learning algorithms, namely, k-Nearest Neighbors, Random Forest and Lasso Regression. Models' performances were evaluated on external test sets derived from the parent datasets either randomly or in a rational manner. We found that many more significant models were obtained when combining 2D and 3D descriptors. We attribute these improvements to the ability of 2D and 3D descriptors to code for different, yet complementary molecular properties.
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
