{"title":"基于多任务深度学习的化学毒性预测协同模型。","authors":"Yuan Yuan Li, Lingfeng Chen, Chengtao Pu, Chengdong Zang, YingChao Yan, Yadong Chen, Yanmin Zhang, Haichun Liu","doi":"10.1002/minf.202200257","DOIUrl":null,"url":null,"abstract":"<p><p>The toxicity of compounds is closely related to the effectiveness and safety of drug development, and accurately predicting the toxicity of compounds is one of the most challenging tasks in medicinal chemistry and pharmacology. In this paper, we construct three types of models for single and multi-tasking based on 2D and 3D descriptors, fingerprints and molecular graphs, and then validate the models with benchmark tests on the Tox21 data challenge. We found that due to the information sharing mechanism of multi-task learning, it could address the imbalance problem of the Tox21 data sets to some extent, and the prediction performance of the multi-task was significantly improved compared with the single task in general. Given the complement of the different molecular representations and modeling algorithms, we attempted to integrate them into a robust Co-Model. Our Co-Model performs well in various evaluation metrics on the test set and also achieves significant performance improvement compared to other models in the literature, which clearly demonstrates its superior predictive power and robustness.</p>","PeriodicalId":18853,"journal":{"name":"Molecular Informatics","volume":"42 5","pages":"e2200257"},"PeriodicalIF":2.8000,"publicationDate":"2023-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Co-model for chemical toxicity prediction based on multi-task deep learning.\",\"authors\":\"Yuan Yuan Li, Lingfeng Chen, Chengtao Pu, Chengdong Zang, YingChao Yan, Yadong Chen, Yanmin Zhang, Haichun Liu\",\"doi\":\"10.1002/minf.202200257\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The toxicity of compounds is closely related to the effectiveness and safety of drug development, and accurately predicting the toxicity of compounds is one of the most challenging tasks in medicinal chemistry and pharmacology. In this paper, we construct three types of models for single and multi-tasking based on 2D and 3D descriptors, fingerprints and molecular graphs, and then validate the models with benchmark tests on the Tox21 data challenge. We found that due to the information sharing mechanism of multi-task learning, it could address the imbalance problem of the Tox21 data sets to some extent, and the prediction performance of the multi-task was significantly improved compared with the single task in general. Given the complement of the different molecular representations and modeling algorithms, we attempted to integrate them into a robust Co-Model. Our Co-Model performs well in various evaluation metrics on the test set and also achieves significant performance improvement compared to other models in the literature, which clearly demonstrates its superior predictive power and robustness.</p>\",\"PeriodicalId\":18853,\"journal\":{\"name\":\"Molecular Informatics\",\"volume\":\"42 5\",\"pages\":\"e2200257\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2023-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Informatics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/minf.202200257\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/minf.202200257","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Co-model for chemical toxicity prediction based on multi-task deep learning.
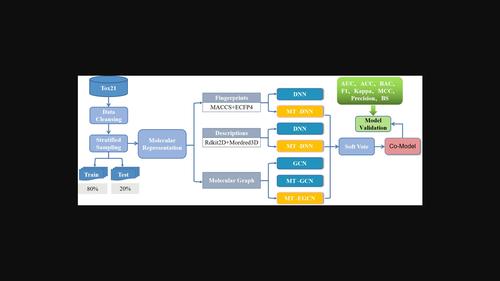
The toxicity of compounds is closely related to the effectiveness and safety of drug development, and accurately predicting the toxicity of compounds is one of the most challenging tasks in medicinal chemistry and pharmacology. In this paper, we construct three types of models for single and multi-tasking based on 2D and 3D descriptors, fingerprints and molecular graphs, and then validate the models with benchmark tests on the Tox21 data challenge. We found that due to the information sharing mechanism of multi-task learning, it could address the imbalance problem of the Tox21 data sets to some extent, and the prediction performance of the multi-task was significantly improved compared with the single task in general. Given the complement of the different molecular representations and modeling algorithms, we attempted to integrate them into a robust Co-Model. Our Co-Model performs well in various evaluation metrics on the test set and also achieves significant performance improvement compared to other models in the literature, which clearly demonstrates its superior predictive power and robustness.
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
